
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom benzodithiophene to diethoxy-benzodithiophene covalent organic frameworks – structural investigations†
Maria S.
Lohse‡
a,
Julian M.
Rotter‡
a,
Johannes T.
Margraf
b,
Veronika
Werner
a,
Matthias
Becker
a,
Simon
Herbert
a,
Paul
Knochel
a,
Timothy
Clark
b,
Thomas
Bein
*a and
Dana D.
Medina
*a
aDepartment of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstraße 5-13, 81377 Munich, Germany. E-mail: bein@lmu.de; dana.medina@cup.uni-muenchen.de
bComputer-Chemie-Centrum, Friedrich-Alexander-University Erlangen-Nürnberg (FAU), Nägelsbachstraße 25, 91052 Erlangen, Germany
First published on 10th March 2016
Abstract
The incorporation of side groups into a covalent organic framework (COF) backbone can be of significant importance for developing new frameworks with enhanced properties. Here we present a novel π-stacked thiophene-based COF containing a benzodithiophene building unit modified with ethoxy side chains. The resulting BDT-OEt COF is a crystalline mesoporous material featuring high surface area and accessible hexagonal pores. We further synthesized a series of COFs containing both BDT and BDT-OEt building units at different ratios. The impact of the gradual incorporation of the BDT-OEt building units into the COF backbone on the crystallinity and porosity was investigated. Furthermore, molecular dynamic simulations shed light on the possible processes governing the COF assembly from molecular building blocks.
Introduction
The self-organization of π-stacked organic layers into highly ordered three-dimensional porous structures endows covalent organic frameworks (2D COFs) with interesting properties for diverse applications, including chemical separations, catalysis and optoelectronics.1–7 The extended organic layers constituting a 2D COF are generally obtained by the copolymerization of rigid building units having complimentary geometries through the formation of strong yet reversible covalent bonds. Boroxines and boronic esters, obtained by condensation reactions of boronic acids and diols, are commonly utilized as integral COF assembly motifs and thereby enable the growth of planar two-dimensional layers.8 Along with the condensation reactions, stacking of the formed organic layers takes place through dispersive forces.9 Therefore, 2D COFs connected through boronate esters feature an assembly of organic building units (or linkers) in a columnar fashion. This permits the formation of supramolecular structures having long-range order, one-dimensional channels and high surface areas.10–15Tuning the structural and physical properties of a COF while maintaining its key features, namely porosity and crystallinity, can pose significant synthetic challenges. For example, incorporating building units having a large conjugated π-system into a framework can be highly desirable for enhanced light absorption properties.11,16 However, such attractive building units tend to exhibit poor solubility under the reaction conditions, often leading to poor crystallinity of the resulting COF.17 To overcome solubility limitations, a modification of the building units with solubilizing agents such as alkyl side chains can be considered. In the final COF structure the solubilizing side chains are oriented orthogonally to the COF pore walls and therefore need to be compatible with the stacking of the COF layers. In addition, these groups can play an important role in defining the electronic properties of the COF. Electron donating or electron withdrawing side groups can have a strong impact on the COF optical bandgap, and they can affect the fine balance between the Coulomb repulsion and the weak attractive London dispersion interactions holding the COF layers together.
In an early study, Lavigne and co-workers reported the synthesis of a boronate ester-linked COF-18 series where the catechol building units were modified with linear alkyl side chains ranging from methyl to propyl groups. The impact of the integrated alkyl side chains on guest molecule uptake was investigated.18 In a later study, enhanced stability towards humidity of the propyl modified COF structure was shown within the COF-18 series.19 However, the overall long-range order of the COF-18 series was significantly diminished with the elongation of the side groups. Jiang and co-workers introduced an azide-modified benzo diboronic acid (BDBA) for the purpose of COF backbone post-modification. In that report, the overall crystallinity of the modified COF-5 was maintained as the azide units having a planar configuration were extended along the plane of the COF layers.20 Dichtel and co-workers introduced an effective crystallization route for boronate ester linked COFs starting from saturated homogenous precursor solutions.9,21 Applying this method, COF structures having high surface area and long-range order were obtained. This crystallization route illustrates the importance of the COF precursor's solubility under reaction conditions.
Recently, we reported the synthesis under solvothermal conditions of oriented thin films of an electron donor COF based on benzodithiophene units, BDT COF.22 We also introduced a novel vapor-assisted conversion approach for the direct deposition of BDT COF films under mild thermal conditions.23
Herein, we describe the synthesis of a highly porous boronate ester linked BDT-OEt COF containing (4,8-diethoxybenzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)diboronic acid. The BDT core is modified with electron-donating ethoxy side chains thereby expanding the family of thiophene-based COFs.7,22,24,25 We synthesized a series of COFs consisting of BDT and BDT-OEt building unit mixtures and studied their structural properties (Scheme 1). The impact of the gradual layer perturbation imposed by the increased fraction of incorporated BDT-OEt building units into the COF backbone on the crystallinity and porosity was investigated. Furthermore, molecular dynamic simulations shed light on the possible processes governing the COF assembly from molecular building units.
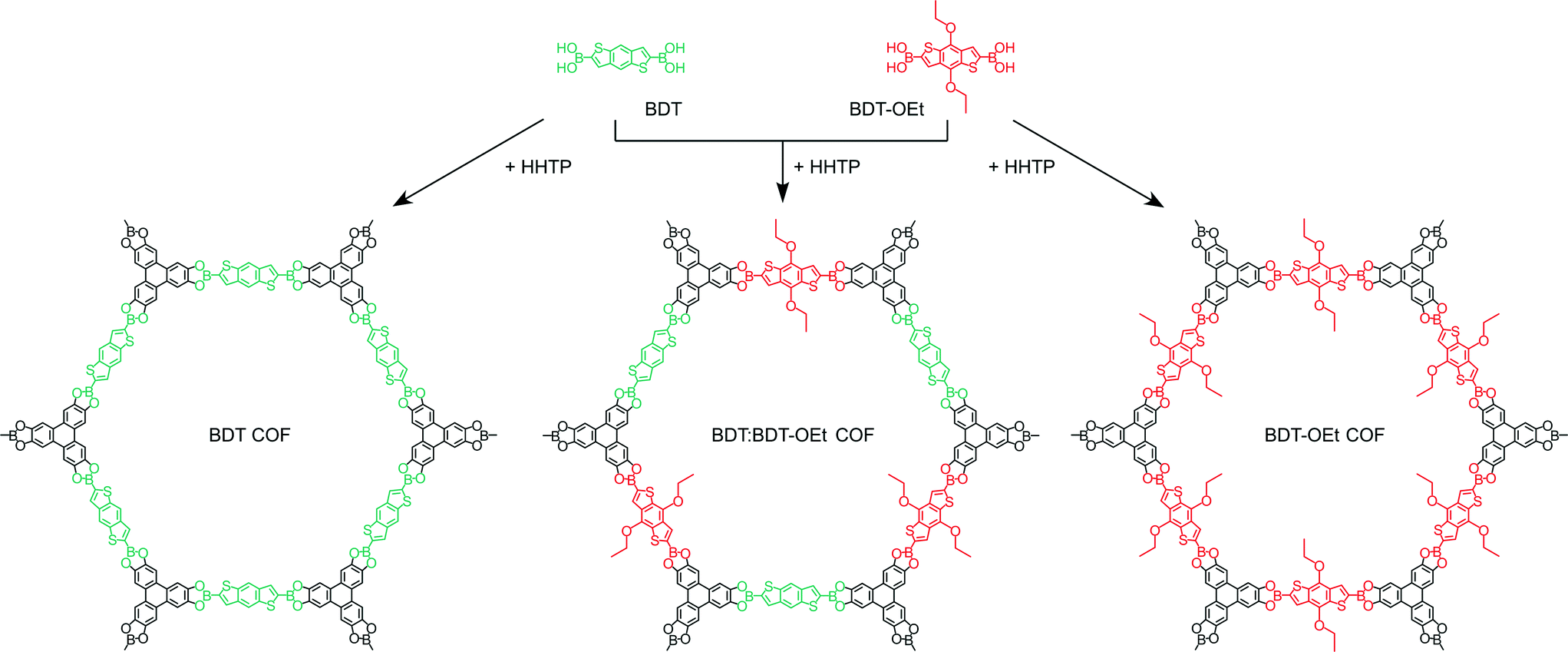 | ||
Scheme 1 Schematic representation of the synthesis of BDT COF (left), BDT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BDT-OEt 1:1 COF (middle), and BDT-OEt COF (right). :BDT-OEt 1:1 COF (middle), and BDT-OEt COF (right). | ||
Experimental
Materials
All materials (if not otherwise noted) were purchased from Aldrich, Fluka, Acros or TCI Europe in the common purities purum and puriss. All materials were used as received without further purification.:1) to afford 1 as a colorless solid (0.675 g, 49%). Mp: 96.9–102.6 °C. 1H NMR (400 MHz, CDCl3) δ/ppm = 7.46 (d, J = 5.5 Hz, 2H), 7.35 (d, J = 5.5 Hz, 2H), 4.35 (q, J = 7.0 Hz, 4H), 1.47 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ/ppm = 144.3, 131.8, 130.4, 126.0, 120.3, 69.4, 16.1. IR (cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3100, 2980, 2873, 1892, 1515, 1477, 1436, 1372, 1347, 1199, 1106, 1085, 1029, 977, 873, 810, 752, 736, 695, 661. MS (70 eV, EI) m/z (%) = 278 (27, M+), 223 (11), 222 (13), 221 (100), 111 (37), 44 (31), 43 (80). HRMS for C14H14O2S2 (278.0435) found: 278.0428.
= 2980, 2927, 2886, 1905, 1520, 1479, 1442, 1370, 1350, 1265, 1170, 1104, 1042, 999, 935, 894, 873, 809, 689, 668. MS (70 eV, EI) m/z (%) = 434 (23, M+), 409 (25), 407 (44), 405 (22), 381 (54), 380 (18), 379 (100), 378 (14), 377 (47), 300 (30), 298 (28), 191 (16), 189 (11), 109 (11), 61 (12), 45 (11), 43 (70). HRMS for C14H12O2Br2S2 (433.8640) found: 433.8649.
:1 THF:diethyl ether mixture (3 × 150 mL). The combined organic layers were dried over Na2SO4. After filtration, the solvents were evaporated in vacuo. The crude product was suspended in isohexane (70 mL) and the mixture was stirred at room temperature for 5 h. The precipitate was filtered off and dried in vacuo.3 was obtained as green solid (184 mg, 50%) and was used without further purification. Mp: 271.9–274.3 °C. 1H NMR (400 MHz, acetone-d6) δ/ppm = 8.14 (s, 2H), 4.39 (q, J = 7.2 Hz, 4H), 1.47 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, acetone-d6) δ/ppm = 145.5, 135.1, 134.5, 130.3, 70.4, 16.8. IR (cm−1): = 3209, 2978, 1532, 1445, 1373, 1344, 1320, 1161, 1084, 1046, 989, 959, 850, 788, 700. HRMS for C14H15O6B2S2 (365.0496) found: 365.0501.
= 3100, 2980, 2873, 1892, 1515, 1477, 1436, 1372, 1347, 1199, 1106, 1085, 1029, 977, 873, 810, 752, 736, 695, 661. MS (70 eV, EI) m/z (%) = 278 (27, M+), 223 (11), 222 (13), 221 (100), 111 (37), 44 (31), 43 (80). HRMS for C14H14O2S2 (278.0435) found: 278.0428.
= 2980, 2927, 2886, 1905, 1520, 1479, 1442, 1370, 1350, 1265, 1170, 1104, 1042, 999, 935, 894, 873, 809, 689, 668. MS (70 eV, EI) m/z (%) = 434 (23, M+), 409 (25), 407 (44), 405 (22), 381 (54), 380 (18), 379 (100), 378 (14), 377 (47), 300 (30), 298 (28), 191 (16), 189 (11), 109 (11), 61 (12), 45 (11), 43 (70). HRMS for C14H12O2Br2S2 (433.8640) found: 433.8649.
:1 THF:diethyl ether mixture (3 × 150 mL). The combined organic layers were dried over Na2SO4. After filtration, the solvents were evaporated in vacuo. The crude product was suspended in isohexane (70 mL) and the mixture was stirred at room temperature for 5 h. The precipitate was filtered off and dried in vacuo.3 was obtained as green solid (184 mg, 50%) and was used without further purification. Mp: 271.9–274.3 °C. 1H NMR (400 MHz, acetone-d6) δ/ppm = 8.14 (s, 2H), 4.39 (q, J = 7.2 Hz, 4H), 1.47 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, acetone-d6) δ/ppm = 145.5, 135.1, 134.5, 130.3, 70.4, 16.8. IR (cm−1): = 3209, 2978, 1532, 1445, 1373, 1344, 1320, 1161, 1084, 1046, 989, 959, 850, 788, 700. HRMS for C14H15O6B2S2 (365.0496) found: 365.0501.
COF synthesis
A 25 mL Schott Duran glass bottle with a Teflon sealed polybutylene terephthalate cap was equipped with 2,3,6,7,10,11-hexahydroxytriphenylene, HHTP (11 mg, 0.034 mmol) and a total of 0.051 mmol of BDT:BDT-OEt linkers in molar ratios of 1:0, 3:1, 1:1, 1:3, and 0:1. The reaction mixture was suspended in 1.5 mL of a 1:1 (v/v) solution of mesitylene/dioxane and heated for 72 h at 100 °C. Afterwards, the green precipitate was isolated by filtration and dried in vacuo for 72 h at 150 °C.
Computational details
The models were defined as periodic supercells containing ten COF layers. MD simulations were performed in the isothermal-isobaric ensemble (at 25 °C and 1 atm), using the Dreiding forcefield as implemented in Forcite Plus and a timestep of 1 fs.27 This forcefield has been shown to provide high quality descriptions of polythiophenes in the condensed phase.28 The average temperature was kept constant using the Andersen thermostat during equilibration and the Nosé–Hoover–Langevin thermostat during production runs. Isotropic pressure was kept constant using the Berendsen barostat. Charges for the periodic COF network were calculated using the QEq scheme.29 Long range electrostatic and van der Waals interactions were treated via the Ewald summation method, with a 6 Å cutoff on the repulsive contribution to the van der Waals energy. In all dynamics runs, the system was equilibrated for 0.1 ns and results were collected over 10 ns. DFT calculations were performed with CASTEP,30 using the PBE functional and the D2 dispersion correction as implemented in the DFT-SEDC code.31–33 We used a plane wave basis set with a 340 eV cutoff and ultrasoft pseudopotentials.34k-point sampling was performed with a 1 × 1 × 6 Monkhorst–Pack grid.35Characterization
Fourier-transform infrared spectra were measured with a Bruker Equinox 55 equipped with a PIKE MIRacle ATR-unit at room temperature in the scan range 650–4000 cm−1. Nitrogen sorption was measured with 20–25 mg sample using a Quantachrome AUTOSORB-1 station at 77.3 K after degassing the sample for at least 12 h under vacuum at 150 °C. The Brunauer–Emmett–Teller (BET) surface areas were calculated from the adsorption branch in the range of p/p0 = 0.05–0.13. Pore sizes were calculated with a QSDFT adsorption model of N2 on carbon (cylindrical, adsorption branch). Powder X-ray diffraction (PXRD) measurements were carried out in reflection mode on a Bruker D8 Discover diffractometer with Ni-filtered Kα-radiation (λ = 1.54060 Å) and a position-sensitive detector (LynxEye). Molecular geometry optimization was performed with Accelrys MS Modeling 4.4 using the universal and Dreiding forcefield method. The final hexagonal unit cell was calculated with the geometric parameters from the optimized structure. For the Pawley refinement and simulation of the PXRD patterns the Reflex module was used (a software package implemented in MS Modeling 4.4). Scanning electron microscopy (SEM) was performed on a JEOL 6500F instrument at an acceleration voltage of 2–5 kV. The samples were coated with a thin carbon layer by carbon fiber flash evaporation at high vacuum. Transmission electron microscopy (TEM) data were obtained with a FEI Titan Themis 60–300 microscope at an acceleration voltage of 300 kV. Cross-polarized 11B and 13C solid-state NMR (ssNMR) measurements were performed on a Bruker Avance III-500 spectrometer in a 4 mm ZrO2 rotor with a spinning rate of 10 kHz. Thermogravimetric analyses (TGA) were recorded on a Netzsch STA 440 C TG/DSC. The measurements proceeded at a heating rate of 10 °C min−1 up to 900 °C, in a stream of synthetic air of about 25 mL min−1. Hydrolyzed COF 1H NMR solution spectra were recorded on a Bruker Avance III-400 MHz spectrometer. 5 mg of COF powder was dissolved in DMSO-d6 containing 5% D2O in every case. A relaxation delay of 8 s was employed to increase the precision of aromatic signal integration.Results
A novel BDT-OEt COF featuring a BDT core decorated with ethoxy side chains was synthesized through the co-condensation of (4,8-diethoxybenzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)diboronic acid and HHTP (2,3,6,7,10,11-hexahydroxytriphenylene) under solvothermal conditions (for more details see Experimental section).Powder X-ray diffraction data of the newly synthesized BDT-OEt COF clearly indicate the formation of a crystalline framework. The diffraction pattern reveals an intensive reflection at 2.78° corresponding to the (100) plane. Additional reflections observed at 4.85°, 5.58° and 7.38°, 9.71° and ∼25.7° (broad) are attributed to the (110), (200), (120), (220) and (001) planes, respectively (Fig. 1a). The obtained d-values correspond to lattice parameters of a = b = 3.66 nm for an ideal hexagonal BDT-OEt COF structure. Effective crystallite domain sizes of 21 nm in diameter for BDT-OEt COF were calculated using the Scherrer equation for reflection broadening. Molecular mechanics simulations for the possible COF layer arrangements were carried out using Materials Studio software and the Forcite module. Assuming a hexagonal symmetry, the BDT-OEt COF unit cell was constructed in the P6 space group corresponding to an ideal eclipsed AA COF layer arrangement, and featuring a completely open pore system (Fig. S1†). To obtain the final unit cell parameters and the crystallite domain sizes, Pawley refinement was employed. For this purpose, the geometry optimized BDT-OEt COF unit cell was refined against the experimental powder X-ray diffraction pattern. Lattice parameters of a = b = 3.65 nm, c = 0.365 nm and crystallite sizes of 20 nm in diameter were obtained. The simulated powder diffraction pattern corrected for reflection broadening is in very good agreement with the experimentally observed pattern with regard to reflection positions and relative intensities (Fig. 1a). Additionally, we simulated the BDT-OEt COF unit cell in the triclinic P1 space group where no symmetry constraints are imposed. After geometry optimization and Pawley refinement, the refined unit cell reveals an eclipsed configuration with tilted COF layers.36 This layer configuration produced a nearly identical simulated diffraction pattern, and therefore the exact eclipsed layer arrangement cannot be determined based on the experimental diffraction pattern (see Fig. S5†). To confirm the eclipsed layer arrangement for the BDT-OEt COF, we also simulated the X-ray diffraction pattern for an alternative hexagonal unit cell with P63 space group corresponding to a staggered, AB layer arrangement (Fig. S8†). Based on the comparison of the simulated AA and AB patterns with the experimentally obtained pattern, we conclude that BDT-OEt crystallizes with an eclipsed layer arrangement allowing for an open pore system.
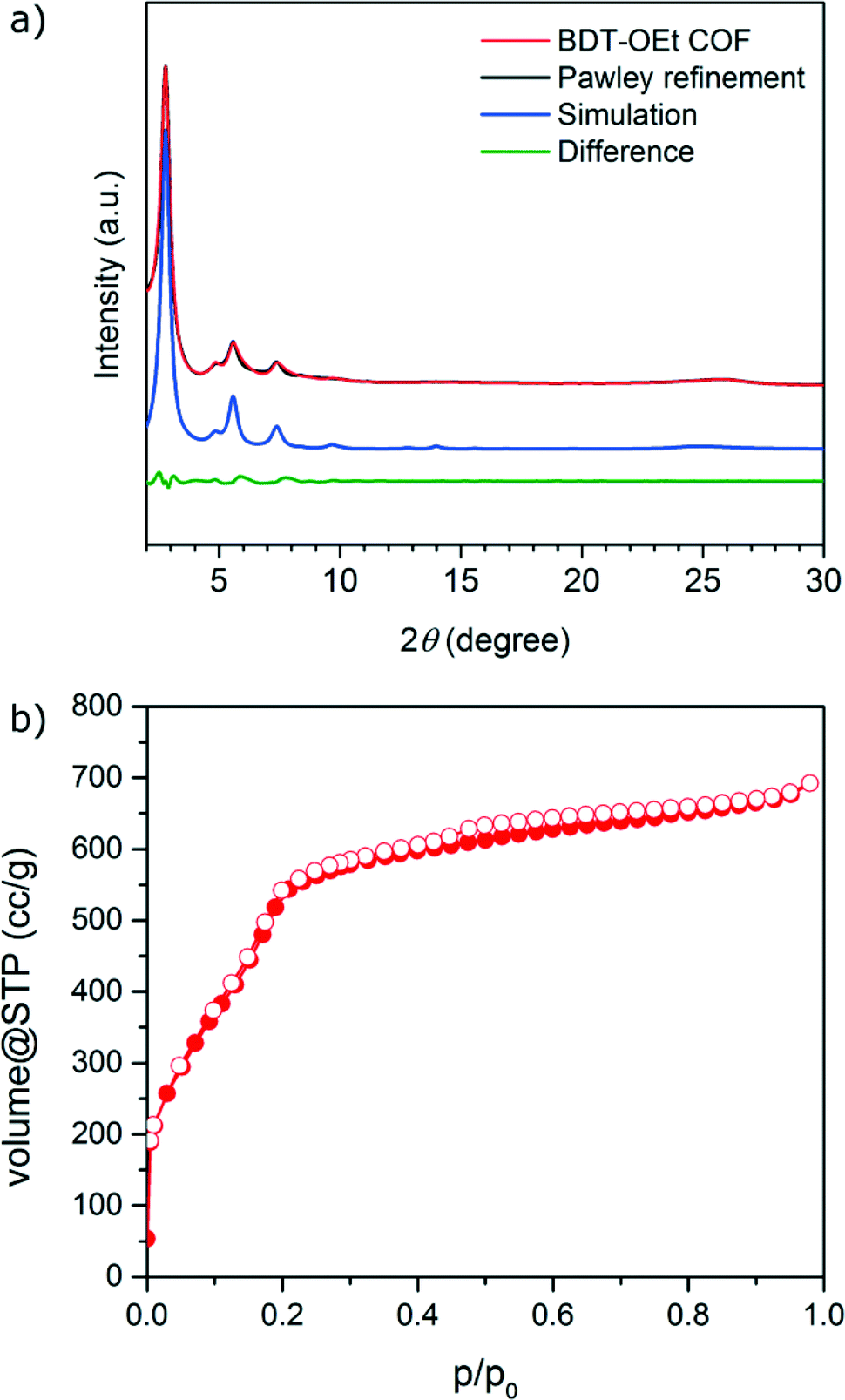 | ||
| Fig. 1 (a) XRD patterns of BDT-OEt COF: experimental (red), Pawley refinement (black), simulated pattern (blue), and difference plot (green). (b) Nitrogen sorption isotherm of BDT-OEt COF. Full symbols: adsorption, empty symbols: desorption. | ||
In previous reports, the side groups decorating the pore walls were considered as an amorphous contribution to the X-ray scattering data, having no preferred orientation in the pore due to their free rotation.18,20,37 To investigate the preferred orientation of the ethoxy groups in the pores, several separate molecular dynamics simulations were carried out (Fig. S10†). Independent of the initial orientation of the groups, which can either face the adjacent hydrogen or sulfur atom of the BDT core or alternate between these two cases, nearly all ethoxy-groups rotated to face the sulfur atom within less than 1 ns. The ethoxy groups retained their position, indicating that the groups can in principle rotate at room temperature, and that the sulfur-facing position is favoured in the crystal (Fig. S10†). This was confirmed by DFT calculations at the PBE+D level, which indicate that the sulfur-facing geometry is more stable by 0.2 eV (corresponding to a Boltzmann population ratio of approx. 4 × 10−4 for the hydrogen-facing geometry at room temperature). This preference can be attributed to the electrostatic attraction between the sulfur lone-pairs and the ethoxy hydrogen atoms.
Nitrogen sorption analysis of an activated BDT-OEt COF powder reveals a type IV sorption isotherm, with a distinct nitrogen uptake at relative low partial pressures, typical for a mesoporous material (Fig. 1b). The Brunauer–Emmett–Teller (BET) surface area was calculated to be as high as 1844 m2 g−1 (p/p0 = 0.05–0.13 in the adsorption branch) and the pore volume to be 1.07 cm3 g−1. These values are in good agreement with the simulated Connolly surface area of 2075 m2 g−1 and calculated pore volume of 1.06 cm3 g−1 for a BDT-OEt COF with an eclipsed layer arrangement. BDT-OEt COF exhibits a narrow pore size distribution as derived from the sorption isotherm with a pore size of 2.9 nm corresponding to a defined mesoporous structure (Fig. S12†).
An aggregation process of COF crystallites into larger domains was discussed as a crucial non-reversible step for the formation of COF precipitates.9 Scanning electron microscopy (SEM) micrographs of BDT-OEt COF bulk material indicate that the small BDT-OEt COF crystallites aggregate into micrometer spherical agglomerates upon COF powder precipitation (Fig. S13†). Transmission electron microscopy (TEM) images reveal an intergrowth of small BDT-OEt COF crystallites into larger polycrystalline domains. In contrast to the BDT COF, less regular fairly short COF channels can be observed (Fig. 2). These observations are in accordance with the reflection broadening observed in the X-ray analysis and suggest that the incorporation of ethoxy side chains can influence the COF growth process in both the ab-plane and the stacking direction.
 | ||
| Fig. 2 Transmission electron microscopy image of BDT-OEt COF showing polycrystalline domains and the COF channels. | ||
Thermogravimetric analysis (TGA) of the COF powder displays a major mass loss at 400 °C that is attributed to framework decomposition (Fig. S19†).
The formation of a boronate ester motif in the COF framework was confirmed by FT-IR and solid-state NMR spectroscopy. IR-bands at 1346 and 1238 cm−1 are attributed to the B–O and C–O vibrational modes of the ester formed (Fig. S20†). In the 11B solid-state NMR spectrum of BDT-OEt COF, a strong peak at 20.9 ppm confirms the presence of a trigonal-planar boron atom of a boronate ester (Fig. S22†). In the 13C solid-state NMR spectrum, the signals at 13.2 ppm and 67.7 ppm are attributed to the methyl and methylene carbon atoms in the ethoxy side chain (Fig. S23†).
Boronate ester linked COFs consisting of building unit mixtures have been introduced in a series of COF-5 structures modified with azide side chains as a platform for a gradual post synthetic modification via click chemistry.20 Herein, we aim at investigating the structural changes imposed by ethoxy groups on a well-defined and highly crystalline COF structure such as the BDT COF. For this purpose, we synthesised a series of COFs where both building units BDT and BDT-OEt are randomly distributed within the COF backbone at different ratios. The BDT:BDT-OEt COF series was synthesised under solvothermal conditions similarly to the synthesis of BDT-OEt COF, with BDT:BDT-OEt building unit ratios of 1:0, 3:1, 1:1, 1:3 and 0:1 (for more information see Experimental section).
Powder X-ray diffraction patterns of the obtained BDT:BDT-OEt COFs confirmed the formation of a series of crystalline frameworks (Fig. 3). Upon the incorporation of ethoxy side chains into the BDT COF backbone, gradual changes in the BDT:BDT-OEt COF diffraction patterns were observed. The relative intensities of the (110) and (200) planes change with the increased fraction of BDT-OEt building units. This is in excellent agreement with the simulated diffraction patterns of BDT and BDT-OEt COFs and further confirms the successful incorporation of BDT-OEt into the frameworks (Fig. S9†). A broadening of the X-ray reflections is evident in BDT-OEt-rich COFs. The effective crystallite domain sizes calculated for BDT:BDT-OEt 3:1 and BDT:BDT-OEt 1:3 COFs are 95 nm and 47 nm, respectively. Therefore, we attribute the reflection broadening to a regular decrease in the crystallite domain sizes with increasing the BDT-OEt building unit content. A gradual small shift of the reflections corresponding to the ab plane to higher 2 theta values with the higher integrated fraction of BDT-OEt building units is observed as well (Fig. S21†). Interestingly, the broad reflection at ∼25.7°, attributed to the layer stacking, maintained its position and its relative intensity. The gradual shift to higher 2 theta values implies that the incorporation of ethoxy groups into the frameworks induced structural modifications expressed in the unit cell parameters. This can be either attributed to a gradual shrinkage of the hexagonal unit cell or to a gradual increase of layer dynamics resulting in a P1 like structure (Fig. S4†).36
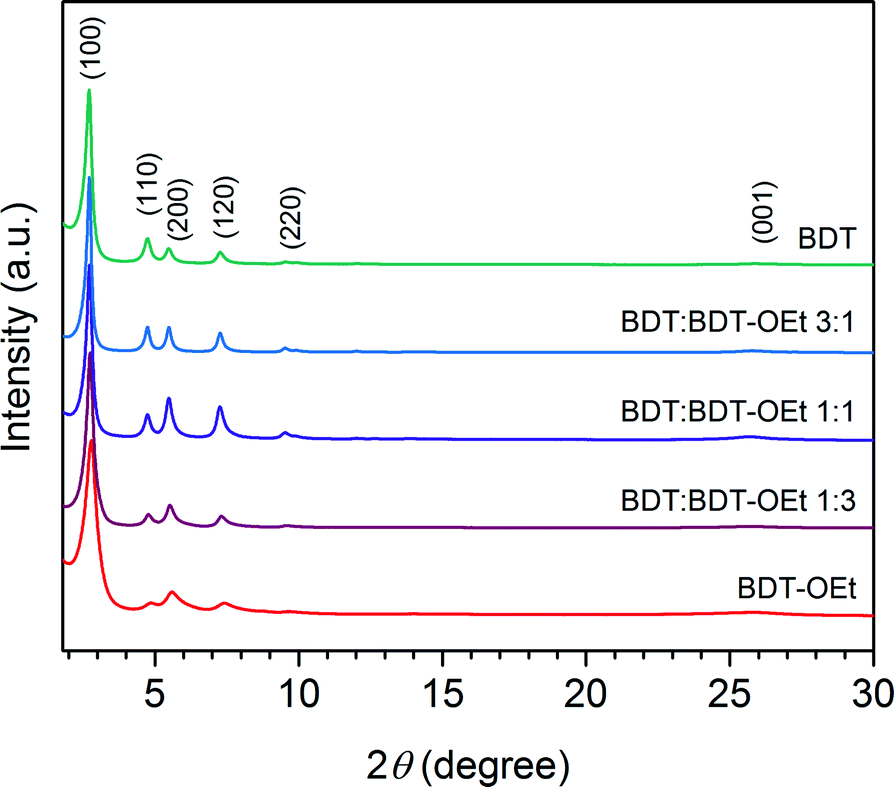 | ||
| Fig. 3 PXRD patterns of BDT COF, BDT:BDT-OEt COFs, and BDT-OEt COF. | ||
Molecular dynamics simulations regarding the layer interactions provided further insights into the effect of the ethoxy groups on the dynamics and order of the system. A comparison of the sulfur–sulfur radial distribution functions gSS(r) for the ethoxy substituted and pristine BDT COF reveals a significant increase in short-range disorder in the former COF (Fig. S11†). This is evident from the broadening and shifted position towards larger distances of the first peak in gSS(r) corresponding to the closest sulfur–sulfur distance between adjacent layers. The increased short-range disorder is attributed mainly to increased movement of the layers in the ab-plane, since more displaced geometries are stabilized by the interactions between the ethoxy groups. Interestingly, the average interlayer distance in the simulations increases when the ethoxy groups are present, which might be expected due to the spatial demands of the ethyl groups. However, this trend regarding the interlayer distances is not observed in the experimental pattern.
The impact of the gradual incorporation of the ethoxy side chains on the pore accessibility of the frameworks was investigated by nitrogen sorption. The BDT:BDT-OEt COF series retained the typical type IV isotherms for mesoporous materials. The relative partial pressure of the slope of the nitrogen uptake is shifted to lower values as expected for the gradually increasing pore occupancy with ethoxy side chains. Pore size distribution evaluation revealed a gradual decrease of the pore size upon the incorporation of ethoxy side chains. In all the BDT:BDT-OEt COF structures only one type of pore is present, indicating a random integration of the ethoxy groups into the frameworks, as opposed to the formation of phase-separated domains. High surface areas were calculated for the BDT:BDT-OEt COF series. This clearly illustrates that incorporation of BDT-OEt building units into the framework does not affect the accessible surface areas and pores of the BDT:BDT-OEt COF (Fig. 4, S12†).
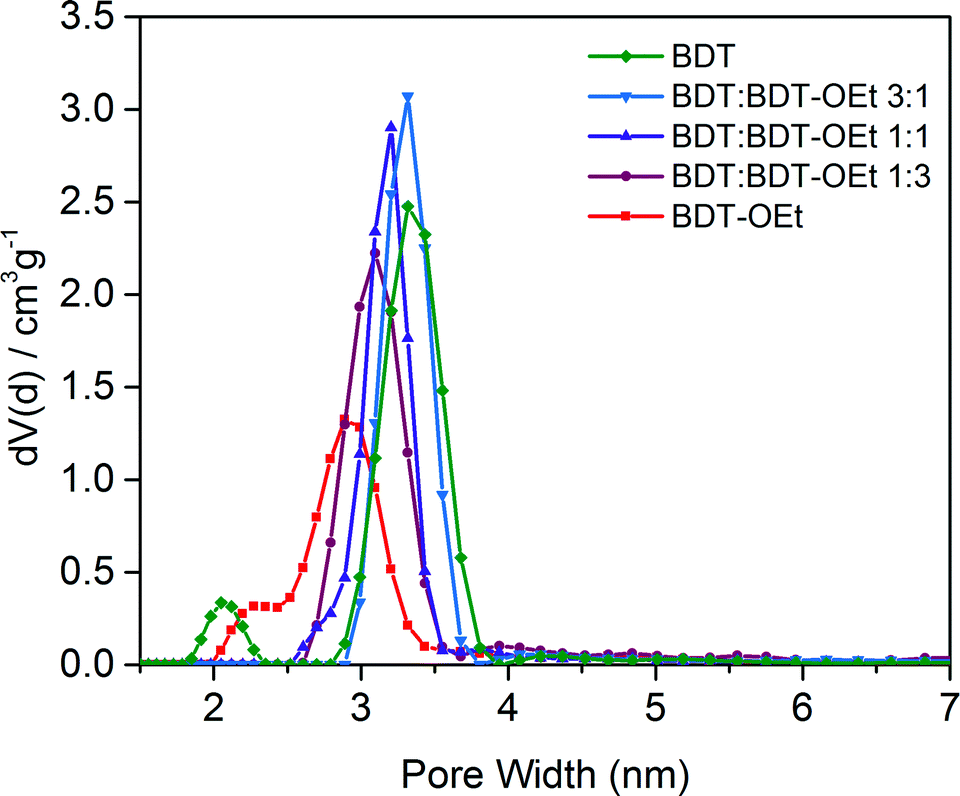 | ||
| Fig. 4 Detail of the pore size distributions of BDT COF, mixed BDT:BDT-OEt COFs and BDT-OEt COF showing the decrease of the main pore diameter upon the incorporation of ethoxy side chains into the COF structure. | ||
The solubility of the respective building units under the reaction conditions plays an important role regarding their incorporation into the COF backbone. If a significant solubility difference would exist, their ratio in the frameworks might deviate from the initial stoichiometric mixture. To determine the final BDT:BDT-OEt ratio incorporated into the frameworks, samples of the respective BDT:BDT-OEt COF series were hydrolysed in wet DMSO-d6 for 1H NMR spectroscopy. Integrating the proton signals of the building units showed that the observed intensities are in very good agreement with the intended stoichiometric ratios of BDT, BDT-OEt and HHTP building units for all COFs. Therefore defects in the form of precursor deficiencies within the frameworks are not observed (Fig. 5 and section 9 in the ESI†).
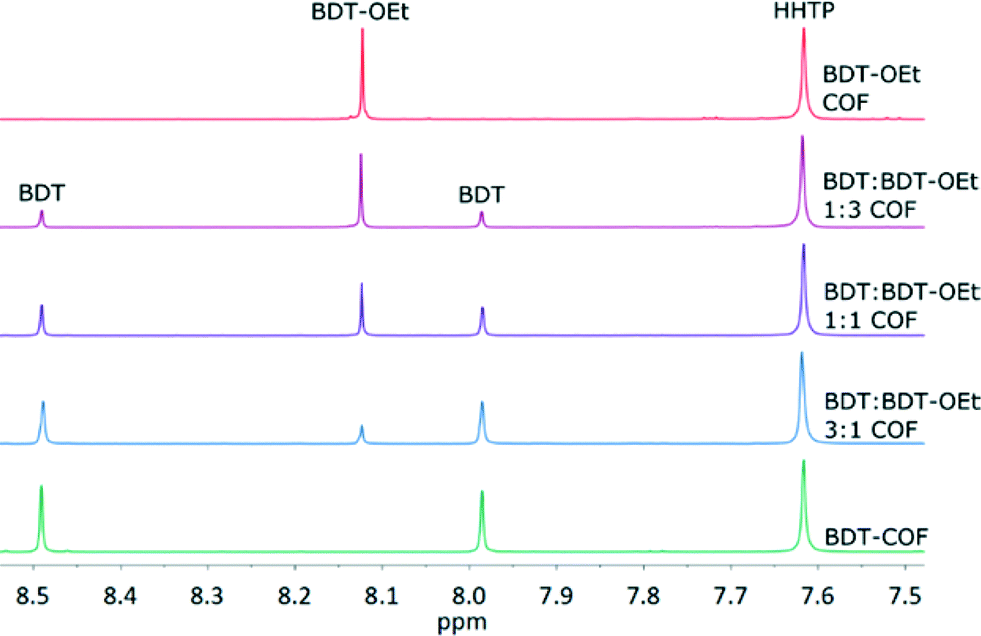 | ||
| Fig. 5 1H-NMR spectra of the hydrolyzed COFs showing the stoichiometric incorporation of BDT and BDT-OEt linkers into the respective frameworks. | ||
SEM micrographs reveal a similar overall morphology for all investigated COFs. Small unevenly shaped particles form large aggregates, comparable with the morphology we observed for BDT-OEt COF (Fig. S13†). In TEM images, BDT-rich COFs show intergrown cauliflower morphology. Depending on the crystallite orientation, ordered channels or the typical honeycomb structure are visible. BDT-OEt-rich COFs feature smaller crystallite domain sizes and shorter COF channels (Fig. S14†). TGA traces were recorded to determine the thermal stability of the COF series. For all COFs, the major mass loss attributed to the decomposition of the framework takes place at 400 °C, showing a similar thermal stability of all investigated frameworks (Fig. S15–S19†).
These analyses confirm that with BDT and BDT-OEt building units, COFs can be formed from freely selected ratios of the respective boronic acids. While mixing these building units in a COF leads to structural changes in the framework, a highly porous, crystalline and thermally stable framework is generated in all cases.
Conclusions
In conclusion, a novel ethoxy-functionalized benzodithiophene based COF, namely BDT-OEt COF, was synthesized under solvothermal conditions. BDT-OEt COF features crystallinity, mesoporosity and high surface area. A series of COFs consisting of both BDT and BDT-OEt building units in different ratios was synthesized. Highly crystalline, mesoporous frameworks were obtained for all BDT:BDT-OEt COFs. A nearly linear evolution from BDT to BDT-OEt COF was observed with regard to pore size and crystallite domain size upon the gradual incorporation of BDT-OEt building units into the COF backbone. Furthermore, we show that the final building unit ratios in the framework can be predetermined prior to the COF synthesis. Therefore, the COF backbones and chemical pore environment can be finely tuned towards desired properties while an eclipsed layer arrangement is retained. Molecular dynamics simulations and DFT calculations shed light on the preferred orientation of the ethoxy groups in the pores. Furthermore, the simulations illustrate that the incorporation of ethoxy chains facilitates a subtle lateral displacement of the BDT COF layers relative to each other.
Acknowledgements
The authors are grateful for funding from the German Science Foundation (DFG; Excellence Cluster NIM) and the Free State of Bavaria (Research Network SolTech). The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement no. 321339. We thank Christian Minke and Thomas Bräuniger for ssNMR, Markus Döblinger for TEM, and Steffen Schmidt and Erika Mühlbauer for SEM measurements.Notes and references
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920–2924 CrossRef CAS PubMed.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- B. J. Smith and W. R. Dichtel, J. Am. Chem. Soc., 2014, 136, 8783–8789 CrossRef CAS PubMed.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS PubMed.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- S. Jin, K. Furukawa, M. Addicoat, L. Chen, S. Takahashi, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505–4511 RSC.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 Search PubMed.
- E. L. Spitler, J. W. Colson, F. J. Uribe-Romo, A. R. Woll, M. R. Giovino, A. Saldivar and W. R. Dichtel, Angew. Chem., Int. Ed., 2012, 51, 2623–2627 CrossRef CAS PubMed.
- H. Yang, Y. Du, S. Wan, G. D. Trahan, Y. Jin and W. Zhang, Chem. Sci., 2015, 6, 4049–4053 RSC.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Löbermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed.
- S. Jin, M. Supur, M. Addicoat, K. Furukawa, L. Chen, T. Nakamura, S. Fukuzumi, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7817–7827 CrossRef CAS PubMed.
- R. W. Tilford, S. J. Mugavero, P. J. Pellechia and J. J. Lavigne, Adv. Mater., 2008, 20, 2741–2746 CrossRef CAS PubMed.
- L. M. Lanni, R. W. Tilford, M. Bharathy and J. J. Lavigne, J. Am. Chem. Soc., 2011, 133, 13975–13983 CrossRef CAS PubMed.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- B. J. Smith, N. Hwang, A. D. Chavez, J. L. Novotney and W. R. Dichtel, Chem. Commun., 2015, 51, 7532–7535 RSC.
- D. D. Medina, V. Werner, F. Auras, R. Tautz, M. Dogru, J. Schuster, S. Linke, M. Döblinger, J. Feldmann, P. Knochel and T. Bein, ACS Nano, 2014, 8, 4042–4052 CrossRef CAS PubMed.
- D. D. Medina, J. M. Rotter, Y. Hu, M. Dogru, V. Werner, F. Auras, J. T. Markiewicz, P. Knochel and T. Bein, J. Am. Chem. Soc., 2015, 137, 1016–1019 CrossRef CAS PubMed.
- G. H. V. Bertrand, V. K. Michaelis, T.-C. Ong, R. G. Griffin and M. Dincă, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4923–4928 CrossRef CAS PubMed.
- S. Duhović and M. Dincă, Chem. Mater., 2015, 27, 5487–5490 CrossRef.
- D. W. Slocum and P. L. Gierer, J. Org. Chem., 1976, 41, 3668–3673 CrossRef CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- O. Alexiadis and V. G. Mavrantzas, Macromolecules, 2013, 46, 2450–2467 CrossRef CAS.
- A. K. Rappe and W. A. Goddard, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS.
- J. Clark Stewart, D. Segall Matthew, J. Pickard Chris, J. Hasnip Phil, I. J. Probert Matt, K. Refson and C. Payne Mike, Z. Kristallogr., 2005, 220, 567 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416–19421 CrossRef CAS PubMed.
- H. Xu, X. Chen, J. Gao, J. Lin, M. Addicoat, S. Irle and D. Jiang, Chem. Commun., 2014, 50, 1292–1294 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: BDT-OEt structure simulations, MD simulations, nitrogen sorption isotherms and pore size distribution calculations; SEM and TEM images; TGA traces; FT-IR spectra; 11B and 13C solid-state NMR spectra of BDT-OEt; 1H NMR spectra of dissolved COFs. See DOI: 10.1039/c6ce00193a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |