
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of Cd(II)/pseudohalide/di-2-pyridyl ketone chemistry – rational synthesis, structural analysis and photoluminescence†
I.
Nawrot
a,
B.
Machura
*a and
R.
Kruszynski
b
aDepartment of Crystallography, Institute of Chemistry, University of Silesia, 9th Szkolna St., 40-006 Katowice, Poland. E-mail: basia@ich.us.edu.pl
bDepartment of X-ray Crystallography and Crystal Chemistry, Institute of General and Ecological Chemistry, Technical University of Lodz, 116 Żeromski St., 90-924 Łódź, Poland. E-mail: rafal.kruszynski@p.lodz.pl
First published on 1st March 2016
Abstract
A systematic investigation of Cd(II)–(py)2CO–X systems (X = N3−, NCO− and NCS−) was conducted, and the following cadmium(II) coordination compounds [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1), [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2), [Cd4(SCN)4{(py)2C(OCH3)(O)}4] (3), [Cd4(N3)4{(py)2C(OCH3)(O)}4] (4), [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5), [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (6), [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7), [Cd4(N3)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (8) and [Cd4(NCO)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (9a and 9b) were successfully synthesized and characterized by single-crystal diffraction, X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC) analysis and IR spectroscopy. The fluorescence properties of 1–9 were studied in the solid state and compared to the fluorescence of di-2-pyridyl ketone. The photoluminescence behaviour of 2–9 was also investigated in acetonitrile solution. The X-ray studies demonstrated a cooperative impact of the organic ligand and auxiliary inorganic ion on the final molecular architectures of the cadmium(II) coordination compounds. Also, the essential roles of intermolecular interactions (hydrogen bonds, π–π stacking interactions and weak O⋯S contacts) in the creation of molecular architectures were discussed.
Introduction
The di-2-pyridyl ketone [(py)2CO] with three potential donor sites is a versatile polydentate ligand whose reactions with transition metal and lanthanide ions have been extensively studied since 1967, when Osborne and McWhinnie reported the first Cu(II) complexes of (py)2CO.1Initially, it was thought that (py)2CO had the ability to coordinate to a metal ion as a tridentate ligand with bonding through the oxygen and two nitrogen atoms or in a bidentate way through the two nitrogen atoms located in the pyridine rings or through one pyridyl nitrogen and the carbonyl oxygen.2
Further studies demonstrated that the ketocarbonyl group of (py)2CO undergoes metal-promoted solvation (H2O, ROH) which yields the gem-diol form (py)2C(OH)2 or hemiketal form (py)2C(OR)(OH) (Scheme 1), coordinating to the metal centres usually in a tridentate way N,N′,O, with the M–O bond often being weak.
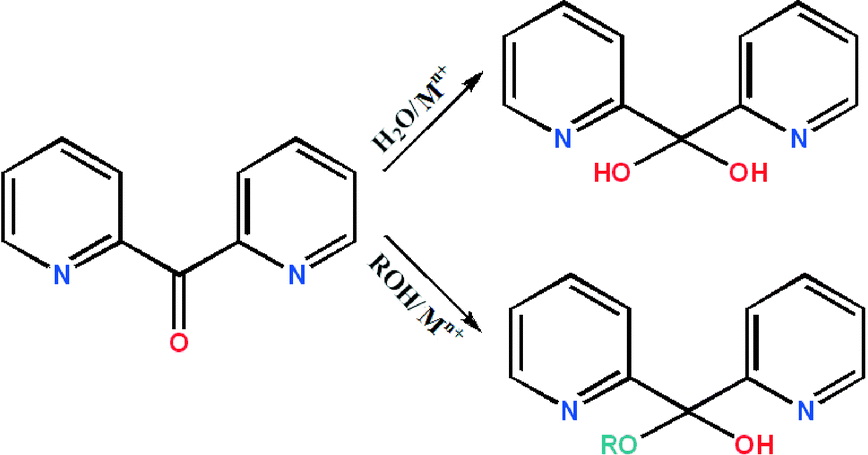 | ||
| Scheme 1 Di-2-pyridyl ketone together with its gem-diol and hemiketal forms. | ||
The solvation process is accounted for the steric strain on the carbonyl carbon produced upon complexation. It is thought that the conversion of the sp2 hybridized C atom in (py)2CO to sp3 (py)2C(OH)2 and (py)2C(OR)(OH) is facilitated by metals in high oxidation states. Complexes with low oxidation state transition metals (Ag+) usually contain the ketone form (py)2CO.3
Interesting coordination modes are also seen when the ligands (py)2C(OH)2 and (py)2C(OR)(OH) become deprotonated. The monoanionic forms usually bridge two (μ2) or three (μ3) metal ions, while the dianionic form can bridge up to five metal sites (μ5) (Scheme 2) yielding various polynuclear compounds of high significance in molecular magnetism and catalysis.4 Among these, special attention has been paid to the cubane-like structures of tetranuclear metal complexes.5
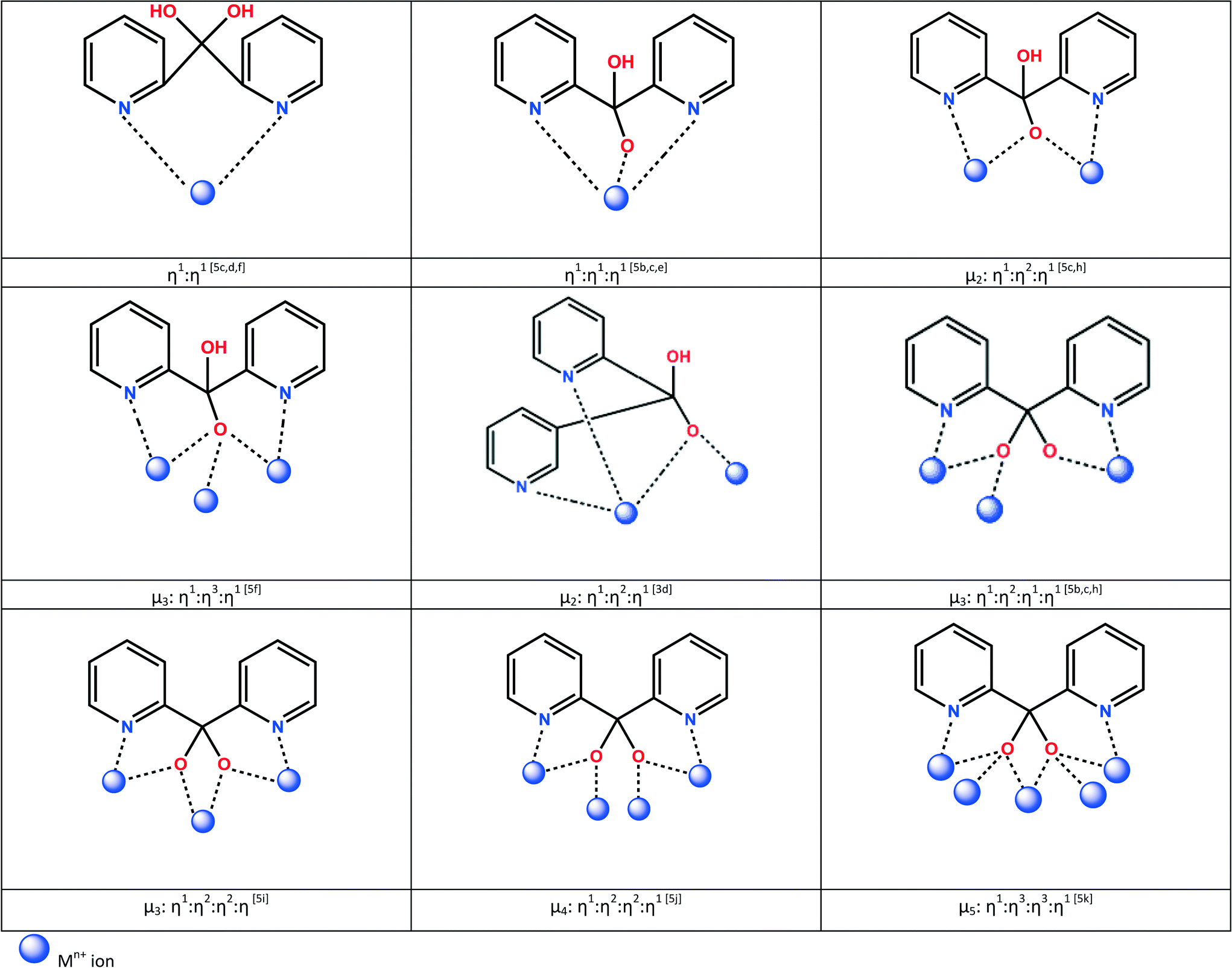 | ||
| Scheme 2 Bridging coordination modes of (py)2C(OH)O− and (py)2CO22−. | ||
Although the use of (py)2CO has been investigated widely in Mn, Fe, Co, Ni, Cu and Zn chemistry,6 its employment in the synthesis of cadmium(II) compounds is rather limited. A search in the CSD (Cambridge Structural Database, version 5.36) revealed only a few Cd(II) compounds incorporating (py)2CO or its derivatives: [Cd2Cl4{(py)2C(OH)(OH)}2]·3H2O,7 [Cd2Br4{(py)2C(OH)(OH)}2]·3H2O,8 [Cd2I4{(py)2C(OMe)(OH)}2],9 [Cd2(NO3)4{(py)2CO}2],5h [Cd4(NO3)4{(py)2C(OH)(O)}4]5h and [Cd4(SCN)4{(py)2C(OMe)(O)}4].5d
Following the well-known strategy of combining the use of heterocyclic organic blockers and pseudohalide ions in the synthesis of coordination compounds, we have conducted a systematic investigation of Cd(II)–(py)2CO–X systems (X = N3−, NCO− and NCS−). By changing the anions, M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :X molar ratio or reaction conditions, we have isolated a series of new pseudohalide Cd(II) compounds [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1), [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2), [Cd4(SCN)4{(py)2C(OCH3)(O)}4] (3), [Cd4(N3)4{(py)2C(OCH3)(O)}4] (4), [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5), [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (6), [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7),
:X molar ratio or reaction conditions, we have isolated a series of new pseudohalide Cd(II) compounds [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1), [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2), [Cd4(SCN)4{(py)2C(OCH3)(O)}4] (3), [Cd4(N3)4{(py)2C(OCH3)(O)}4] (4), [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5), [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (6), [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7),
[Cd4(N3)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (8) and [Cd4(NCO)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (9a and 9b), which were structurally characterized by single crystal X-ray diffraction analysis and further characterized by infrared spectroscopy (IR), elemental analyses, X-ray powder diffraction (XRPD) and differential scanning calorimetry (DSC) analyses. The X-ray studies demonstrated a cooperative impact of the organic ligand and auxiliary inorganic ion on the final molecular architectures of cadmium(II) coordination compounds. An essential role of intermolecular interactions (hydrogen bonds, π–π stacking interactions and weak O⋯S contacts) in the creation of molecular architectures was also proven. The luminescence properties of di-2-pyridyl ketone and cadmium(II) coordination compounds were studied in solution and the solid state.
Experimental section
Synthetic procedures
390) and 235 (98340).
550).
650) and 233 (69460).
110).
120).
006).
391).
 | ||
| Scheme 3 Crystal forms of 9a and 9b. | ||
Physical measurements
IR spectra were recorded on a Nicolet iS5 FT-IR spectrophotometer in the spectral range of 4000–400 cm−1 with the samples in the form of KBr pellets. Absorption UV–vis spectra were recorded on a UV–vis-NIR Nicolet iS50 double beam spectrophotometer. The steady-state and time-resolved emission spectra were measured for the solid state and MeCN solutions in ambient temperature on a FLS-980 fluorescence spectrophotometer equipped with a 450 W Xe lamp and a high-gain photomultiplier PMT+500nm (Hamamatsu, R928P) detector. The PL lifetime measurement was performed with a time correlated single photon counting (TCSPC) method. The excitation wavelength (340, 370 and 470 nm) was obtained using a TCSPC diode with a 100 ns pulse period as a light source and a PMT was used as a detector. IRF was designated using sample (solid state measurement) or a Ludox solution as a standard. The quantum yields of fluorescence were determined using an absolute method in room temperature, using an integrating sphere with the solvent as a blank.X-ray powder diffraction (XRPD) measurements were performed on a PANalytical Empyrean X-ray diffractometer using Cu-Kα radiation (λ = 1.5418 Å) or Co-Kα radiation (λ = 1.789 Å), in which the X-ray tubes were operated at 40 kV and 30 mA ranging from 5 to 80°.
The DSC measurements were made with the DSC 200 F3 Maia differential scanning calorimeter (NETZSCH). The measurements were performed under a flow of analytical grade nitrogen (flow rate of 20.0 cm3 min−1) in the temperature range of 293.16–673.16 K. Aluminum crucibles were used with a mass difference smaller than 0.005 mg and a heating rate 20.0 K min−1 was applied. The specific heat capacity of all crucibles was tested before measurements and the difference between the reference crucible and the sample containing crucible was not larger than 0.01 J g−1. The temperature and enthalpy were calibrated with usage of six spectrally pure standards: adamantane, In, Sn, Bi, Zn and CsCl. The sample mass was 3.02, 2.33, 1.53, 1.50, 3.00, 5.09, 1.96 and 3.25 mg for 1, 2, 4, 5, 6, 7, 8 and 9a and 9b, respectively.
Crystal structure determination and refinement
The X-ray intensity data of 1–9 were collected on an Gemini A Ultra Oxford Diffraction four-circle kappa geometry diffractometer with an Atlas CCD detector and graphite monochromatic MoKα radiation (λ = 0.71073 Å) at 293.0(2) K. The unit cell determination and data integration were carried out using the CrysAlis package of Oxford Diffraction.10 Intensity data were corrected for the Lorentz and polarization effects. The absorption correction was introduced by SCALE3 ABSPACK scaling algorithm.10 The structures were solved by the Patterson method using SHELXS-97 and refined by full matrix least-squares on F2 with SHELXL-97 with anisotropic displacement parameters for non-hydrogen atoms.11 Hydrogen atoms were placed in calculated positions refined using idealized geometries (riding model) and assigned fixed isotropic displacement parameters, d(C–H) = 0.93 Å, Uiso(H) = 1.2 Ueq(C) (for aromatic); and d(C–H) = 0.96 Å, Uiso(H) = 1.5 Ueq(C) (for methyl). The methyl groups were allowed to rotate about their local threefold axis. Details concerning crystal data and refinement are summarized in Tables S1 and S2.† The hydrogen bonds and selected bond distances and angles of 1–9 are listed in Tables S3–S12 (ESI†).Results and discussion
Synthesis and general characterization
The compounds [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1), [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2), [Cd4(SCN)4{(py)2C(OCH3)(O)}4] (3), [Cd4(N3)4{(py)2C(OCH3)(O)}4] (4), [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5), [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (6), [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7), [Cd4(N3)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (8) and [Cd4(NCO)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (9a and 9b) have been synthesized via a self-assembly process by reacting NH4SCN, NaN3 or NaCNO with Cd(NO3)2 and the heterocyclic ligand di-2-pyridyl ketone [(py)2CO]. In all the reactions the metal-to-organic ligand ratio was equal, 1:1. The synthetic strategy for 1–9 is presented in Scheme 4.
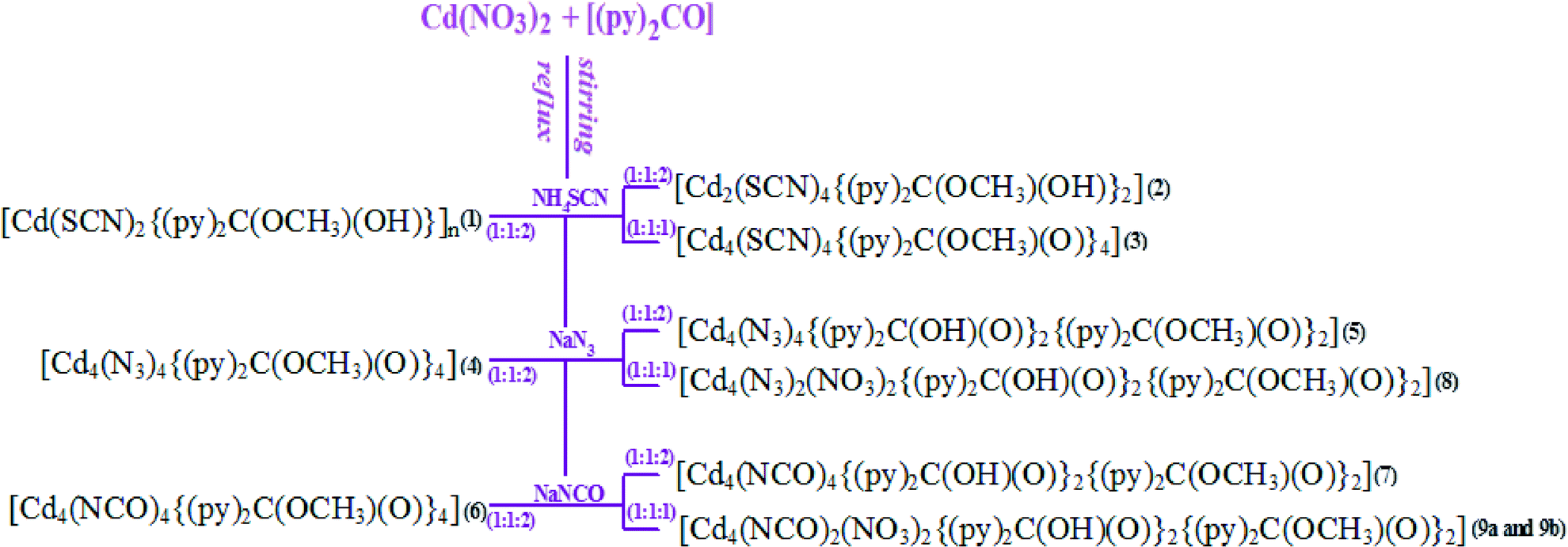 | ||
| Scheme 4 The synthetic strategy for 1–9. | ||
As it can be seen in Scheme 4, the pseudohalide ligand and reaction variables such as the molar ratio of metal precursor to pseudohalide anions and temperature strongly influence the structural versatility of the Cd(II)–(py)2CO–X systems. The 1:1:2 reactions between Cd(NO3)2·4H2O, di-2-pyridyl ketone and NaN3 or NaNCO in MeOH yielded tetranuclear compounds: [Cd4(X)4{(py)2C(OCH3)(O)}4] (X = N3− in 4 and NCO− in 6) – when the syntheses were carried out under reflux, and [Cd4(X)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] X = N3− in 5 and NCO− in 7) – at room temperature. In contrast, the 1:1:2 reactions of Cd(NO3)2·4H2O, (py)2CO and NH4SCN resulted in the formation of one-dimensional coordination polymer [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1) – under reflux, and [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2) – at room temperature. A tetranuclear compound [Cd4(SCN)4{(py)2C(OMe)(O)}4] was isolated by reacting Cd(NO3)2·4H2O, (py)2CO and NH4SCN in a 1:1:1 molar ratio. Many attempts were made to obtain the tetranuclear compound of formula [Cd4(SCN)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2], but all the efforts were in vain. The use of a 1:1:1 molar ratio of Cd(NO3)2·4H2O, (py)2CO and NaN3 or NaNCO in MeOH at room temperature led to the formation of [Cd4(X)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2]. In all cases, the metal-promoted solvation of the ketocarbonyl group of (py)2CO occurs and the formation of the gem-diol form (py)2C(OH)2 or hemiketal form (py)2C(OCH3)(OH) is observed. Depending on the type of pseudohalide ligand and reaction conditions, the gem-diol, hemiketal or their deprotonated forms (Scheme 4) coordinate to the metal center in a tridentate way N,N′,O.
The phase purity of the complexes was verified by PXRD measurements. As shown in Fig. S1–S8 (ESI†), the XRPD patterns measured for the polycrystalline samples were in good agreement with the XRPD patterns simulated from the respective single-crystal X-ray data using the Mercury 2.4 program,12 demonstrating that the crystal structures are truly representative of the bulk materials.
In the high frequency region (3460–3100 cm−1), the IR spectra of 1, 2, 5 and 7–9 show broad absorptions, assignable to O–H stretching vibrations. Intense absorptions associated with the stretching modes of SCN− ions occur at 2117 and 2084 cm−1 for 1, 2126 and 2100 cm−1 for 2, and 2094 cm−1 for 3. The occurrence of two strong absorptions for 1 and 2 is in agreement with the two different coordination modes of the thiocyanate groups in these structures. The stretching vibrations of N3− ions are seen as strong as very strong absorptions at 2048 cm−1 for 4, 2064 and 2036 cm−1 for 5 and 2070 cm−1 for 8, whereas the stretching modes νa(NCO) occur at 2174 cm−1 for 6, 2170 cm−1 for 7, and 2173 cm−1 for 9. The conversion of the sp2 hybridized C atom in free (py)2CO to sp3 in (py)2C(OH)2, (py)2C(OR)(OH) and their deprotonated forms 1–9 is evidenced by the disappearance of the absorption band associated with the carbonyl group (at 1683 cm−1 for free (py)2CO) and enhancement of the band at ∼1040 cm−1 attributed to the stretching vibration of the C–O bond.13 Finally, the spectrum of the mixture of 9a and 9b does not contain any obvious splitting and duplication of the bands, which proves that the oscillators of both compounds have a similar strength (Fig. S9–S16 in the ESI†).
The heating (under a nitrogen atmosphere) of the compounds containing the bridging μ1,3-SCN ions (1 and 2) or μ1,1-NCO ones (with the exception of 9) leads to melting of the compounds, at about 400 K (1 and 2) and 480 K (3) directly followed by the decomposition (starting at about 430 K (1 and 2) and 510 K (3)). The melting of compounds 1 and 2 is a fast process exhibiting as the strong endothermic peak on the DSC curve (about 30 K wide), while the melting of compound 7 is a slow transformation (displayed by a broad endothermic peak with four overlapping, but distinguishable maxima). The similarity of the processes observed for the compounds 1 and 2 originates from the analogous linkage of the same anion (SCN−) to the metal centres, and the dissimilarity of the effect observed for 7 (accompanied by higher thermal stability) is caused by the subsequent dissociation of the coordination bonds formed by the NCO ion (the μ1,1 coordination versus μ1,3 coordination). The final decomposition processes start above 550 K and lead to elemental carbon as a dominant solid product (confirmed by the XRPD diffraction of the sinter) in the cases of 1 and 2. Compound 7 retains some of the initial composition, as it is observed during cooling of the samples (the exothermic crystallisation in the temperature range of 588–579 K, with an enthalpy of −9.2 J g−1(residue)).
The heating of compounds possessing solely μ1,1-NCO or μ1,1-N3 bridges or nitrate ions (4–6, 8 and 9) leads in each case to a vigorous exothermic decomposition process (with an enthalpy larger than −280 kJ mol−1) starting at almost 500 K, and preceded by a very small endothermic effect (with an enthalpy about 10 kJ mol−1), associated with the formation of the quasi-liquid films on the sample surfaces. Further elevation of temperature leads to subsequent, overlapping stages of thermal decomposition of formed transition products. These processes lead to elemental carbon as a dominant solid product. The high thermal stability of these compounds (about 100 K larger than that of compounds 1, 2, and 7) originates from the formation of the stable coordination core via bridging atoms. The observed difference in thermal stability and behaviour between 7 and 9 may be attributed to the presence of the chelating NO3− ions (the chelation in 9 increases the stability in comparison to the terminal binding of two monodentate NCO− ions of 7, and the oxidative properties of NO3− ions favour decomposition over melting). The DSC analysis of the compounds 9a and 9b does not show the presence of the two different polymorphs of the compounds (the first extremum exhibits the regular sharp shape), even in the case of the study of the evident mixture of both polymorphs (determined via the XRPD). This effect originates from the character of the first process. Typically the polymorphic forms differ in the crystal lattice energy as a result of the different characters of intermolecular interactions in the crystal net. Consequently the polymorphs possess different enthalpies of fusion. Compound 9 decomposes without melting, which means that the energetic effects are molecular-structure related, and they are not governed by the crystal structure. Regardless of the process character (melting or decomposition), 9a and 9b do not form any strong or medium strength hydrogen bonds, thus the energetic effects related to the degradation of the crystal net are negligible (see also Fig. S17–S24†).
Structure of [Cd(NCS)2{(py)2C(OCH3)(OH)}]n (1)
Compound 1 crystallizes in the chiral orthorhombic space group P212121. Single-crystal X-ray diffraction analysis was performed for eight crystals, randomly picked from the same reaction. The Flack parameters of the eight crystals were all close to zero (Tables S1 and S2†), indicating a large degree of enantiomeric purity within the crystal.14 Furthermore, the results demonstrated that the bulk material contained only single crystals of M helices.The X-ray crystal structure analysis revealed that the cadmium(II) centres in 1 are singly bridged by end-to-end thiocyanate ions with the formation of an infinite left-handed helical chain, coincident with the 21 screw axes along the a crystal direction and with a pitch of 8.9163(4) Å which is equal to the b axis length (Fig. 1). The intrachain Cd⋯Cd separation of 6.181(4) Å is in accordance with the values reported for related cadmium(II) structures based on single thiocyanate bridges, 6.006 Å in [Cd3(tren)2(SCN)6]n,15 6.232 Å in [Cd(ben)(SCN)2)] (ben is bis(2-aminoethyl)amine)16 and 6.200 Å in [Cd3(NCS)6(medien)2]n·nH2O (medien is N,N-bis(2-aminoethyl)methylamine).17
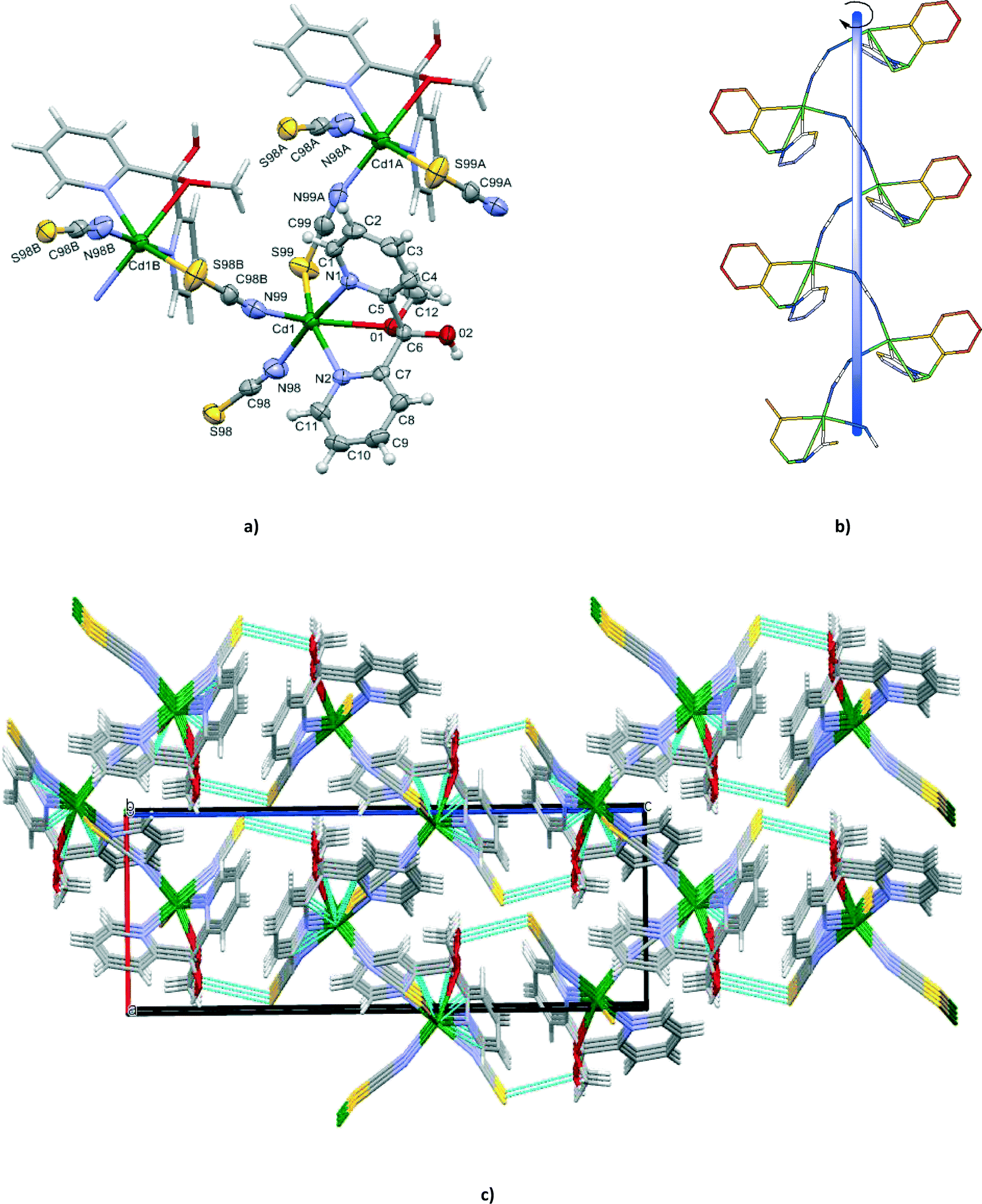 | ||
| Fig. 1 (a) Perspective view of the fragment of [Cd(SCN)2{(py)2C(OCH3)(OH)}]n (1) showing the atom numbering. Displacement ellipsoids are drawn at 50% probability. Symmetry codes: (A) = x + 1/2, −y + 1/2, −z + 1, (B) = x − 1/2, −y + 1/2, −z + 1; (b) perspective view of M helix; (c) view of the three dimensional supramolecular network formed via a weak interaction O(2)⋯S(99)i [(i): 2 − x, 0.5 + y, 1.5 − z) interactions shown by dotted lines. | ||
Each cadmium(II) centre is in a distorted {N4OS} octahedral coordination environment defined by two nitrogen and one oxygen atoms from the hemiketal form (py)2C(OCH3)(OH), terminal N-coordinated thiocyanate anion and two μ1,3-SCN− ions, one bounded by N and one by the S donor. The tridentate N,N′,O(CH3) coordination mode produces a six-membered metallocyclic ring (Cd(1)–N(1)–C(5)–C(6)–C(7)–N(2)) and two five-membered (Cd(1)–N(1)–C(5)–C(6)–O(1) and Cd(1)–N(2)–C(7)–C(6)–O(1)) metallocyclic rings fused along the Cd(1)–O(1)–C(6). The pyridyl rings of the (py)2C(OCH3)(OH) ligand are planar and form a dihedral angle of 67.76(15)°. The two five-membered chelating rings Cd(1)–N(1)–C(5)–C(6)–O(1) and Cd(1)–N(2)–C(7)–C(6)–O(1) exist in the envelope form where the O(1) atom is 0.986(5) and 1.022(4) Å out of the plane of the remaining four atoms, respectively. The six-membered metallocyclic ring (Cd(1)–N(1)–C(5)–C(6)–C(7)–N(2)) is in a boat conformation with the Cd(1) and C(6) atoms being 0.964(6) and 0.791(6) Å, respectively, out of the best mean plane defined by the N(1), N(2), C(5) and C(7) atoms. The pyridyl nitrogens can be viewed as strongly coordinating to the metal (2.322(3) and 2.402(3) Å), while the methylated oxygen forms a weak bond to Cd(II) (2.603(3) Å).
The bridging and terminal thiocyanate units are quasi-linear as reflected in their bond angles of 177.3(4) and 179.0(5)°, respectively. Substantial bending occurs at the sulphur atom of the thiocyanate ion [C(99)i–S(99)–Cd(1) = 101.84(16)°; i: x − 1/2, −y + 1/2, −z + 1].
The helical chains are assembled into a three-dimensional supramolecular framework (Fig. 1c) by a weak O(2)⋯S(99)i interaction [(i): 2 − x, 0.5 + y, 1.5 − z] with a distance of 3.228 Å, which is only marginally shorter than the sum of the van der Waals radii (3.3 Å).18
Structure of [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2)
The structure of 2 consists of the dinuclear [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] units as presented in Fig. 2a. The dimer results from the pairing of the two mononuclear units [Cd(SCN)2{(py)2C(OCH3)(OH)}], related by a crystallographic center of inversion. The Cd(II) ions are doubly bridged by thiocyanate anions in an end-to-end coordination mode. The double NCS− bridge forms an essentially planar, rectangular eight-membered Cd(μ1,3-SCN)2Cd ring with an intradinuclear Cd⋯Cd separation of 5.4826(6)Å, slightly shorter in comparison with the related dinuclear cadmium(II) coordination compounds based on a double thiocyanate bridge and incorporating tridentate terminal co-ligand: 5.889 Å in [Cd2(C11H14N3O3)2(μ1,3-SCN)2(CH4O)2],19 5.659 Å in [Cd2(pydim)2(μ1,3-SCN)]2[BF4]2,20 5.791 Å in [Cd(L1)(NCS)(μ1,3-SCN)]2 (L1 = N-(4,6-dimethyl-pyrimidin-2-yl)-N′-pyridin-2-ylmethylene-hydrazine),21 and 5.887 Å in [Cd(L2)(NCS)(μ1,3-SCN)]2 (L2 = N,N-diethyl-N′-(1-pyridine-2-yl-ethylidene)-ethane-1,2-diamine).22 | ||
| Fig. 2 (a) Molecular structure of [Cd2(SCN)4{(py)2C(OCH3)(OH)}2] (2) showing the atom numbering. Displacement ellipsoids are drawn at 50% probability. Symmetry codes: (A) = −x, −y + 1, −z + 1; (b) view of the packing of 2 showing the hydrogen bonds O(2)–H(2A)⋯N(98)i [D⋯A distance = 2.769(7) Å and D–H⋯A angle = 167.0°; (i): −x, 2 − y, 1 − z]. | ||
Each cadmium(II) ion is six-coordinated in a highly distorted {N3S2O} octahedral geometry defined by two nitrogen and one oxygen atoms of the tridentate chelating (2-py)2C(OCH3)(OH) molecule, sulphur and nitrogen atoms from the bridging thiocyanate ligands and a sulphur atom from the terminally bound thiocyanate ion. The significant distortion from an ideal octahedron can be attributed to the N,N′,O-tridentate coordination of (py)2C(OCH3)(OH) which elongated the M–O bond length (2.498(3) Å) as well as the length of the Cd–S distances (2.6336(14) and 2.5953(15) Å). Likewise in 2, the six-membered chelate ring Cd(1)–N(1)–C(5)–C(6)–C(7)–N(2) adopts a boat conformation with the Cd(1) and C(6) atoms on the same side of the plane formed by the other four atoms. The dihedral angle between the mean planes of the two coordinated pyridyl rings is 68.86(18)°, very close to the value found in 1. The bridging and terminal thiocyanate units are quasi-linear, as reflected in their bond angles of 178.4(4) and 177.9(6)°, respectively. Substantial bending occurs at the sulphur atom of the thiocyanate ions [C(98)–S(98)–Cd(1) = 99.45(18)° and C(99)–S(99)–Cd(1) = 92.58(17)°] and also at the nitrogen atom of the thiocyanate bridge [C(99)–N(99)–Cd(1)i = 159.9(4)°; i: −x, −y + 1, −z + 1].
The neighbouring molecules [Cd2(SCN)4{(py)2C(OCH3)(OH)} are linked through the O(2)–H(2 A)⋯N(98)i hydrogen bonds involving the uncoordinated hydroxy oxygen of the (py)2C(OCH3)(OH) and the nitrogen atom of the terminal thiocyanate ligand [D⋯A distance = 2.769(7) Å and D–H⋯A angle = 167.0°; (i): −x, 2 − y, 1 − z] into a 1D supermolecular chain network (Fig. 2b).
Structures of [Cd4(N3)4{(py)2C(OCH3)(O)}4] (4) and [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (6)
The compounds 4 and 6 crystallize in the tetragonal space group I41/acd and their crystal structures consist of tetranuclear molecules [Cd4(N3)4{(py)2C(OCH3)(O)}4] (in 4) and [Cd4(N3)4{(py)2C(OCH3)(O)}4] (in 6) with cubane-like [Cd4(μ3-O)4]4+ cores. The vertices of the cubane structures are alternately occupied by four Cd atoms and four oxygen atoms of monoanion (py)2C(OCH3)O−, symmetrically related by a crystallographic four-fold inversion axis symmetry (Fig. 3). In addition to three μ3 oxygen atoms, each Cd(II) center is coordinated to two N pyridine donors belonging to two different (py)2C(OCH3)O− ligands and one terminal pseudohalide ion (N3− in 4 and NCO− in 6) to complete an octahedral coordination environment. Each monoanion (py)2C(OCH3)O− coordinates to three Cd(II) centers forming two five-membered chelating rings with two different metals and an alkoxide type bond to a third Cd(II) atom. The dihedral angle between the mean planes of the pyridyl rings belonging to the same (py)2C(OCH3)O− ligand is 79.87° in 4 and 81.18° in 6. Upon coordination, the monoanion (py)2C(OCH3)O− forms one short (2.246(4) Å in 4 and 2.248(5) Å in 6) and two elongated (2.392(3) and 2.449(4) Å in 4, 2.363(5) and 2.248(5) Å in 6) Cd–O bond lengths. The different Cd–O bond lengths are reflected in the Cd⋯Cd separations; the face diagonal vectors Cd⋯Cd are 3.6502(8) and 3.6617(8) Å in 4 and 3.6515(8) and 3.6603(8) Å in 6. The cube clearly deviates from the ideal geometry. The internal cube angles at the metal vertices have values 79.37(13) 76.33(14) and 75.17(13)° in 4 and 79.50(18) 75.11(18) and 77.52(19)° in 6, whereas the comparable angles at the alkoxide corners are larger than 90°, 97.90(13), 102.49(14) and 104.31(14)° in 4 and 98.12(17), 100.98(19) and 104.69(19)° in 6. The pyridyl rings are involved in π–π interactions; 3.636(3) Å for Cg(23)i((N(1)–C(1)–C(5))⋯Cg(23)((N(1)–C(1)–C(5)) centroids for 4 and 3.613(5) Å for Cg(24)i((N(2)–C(7)–C(11))⋯Cg(23)((N(2)–C(7)–C(11)) centroids for 6 (i) = 1 − x, 1/2 − y, z.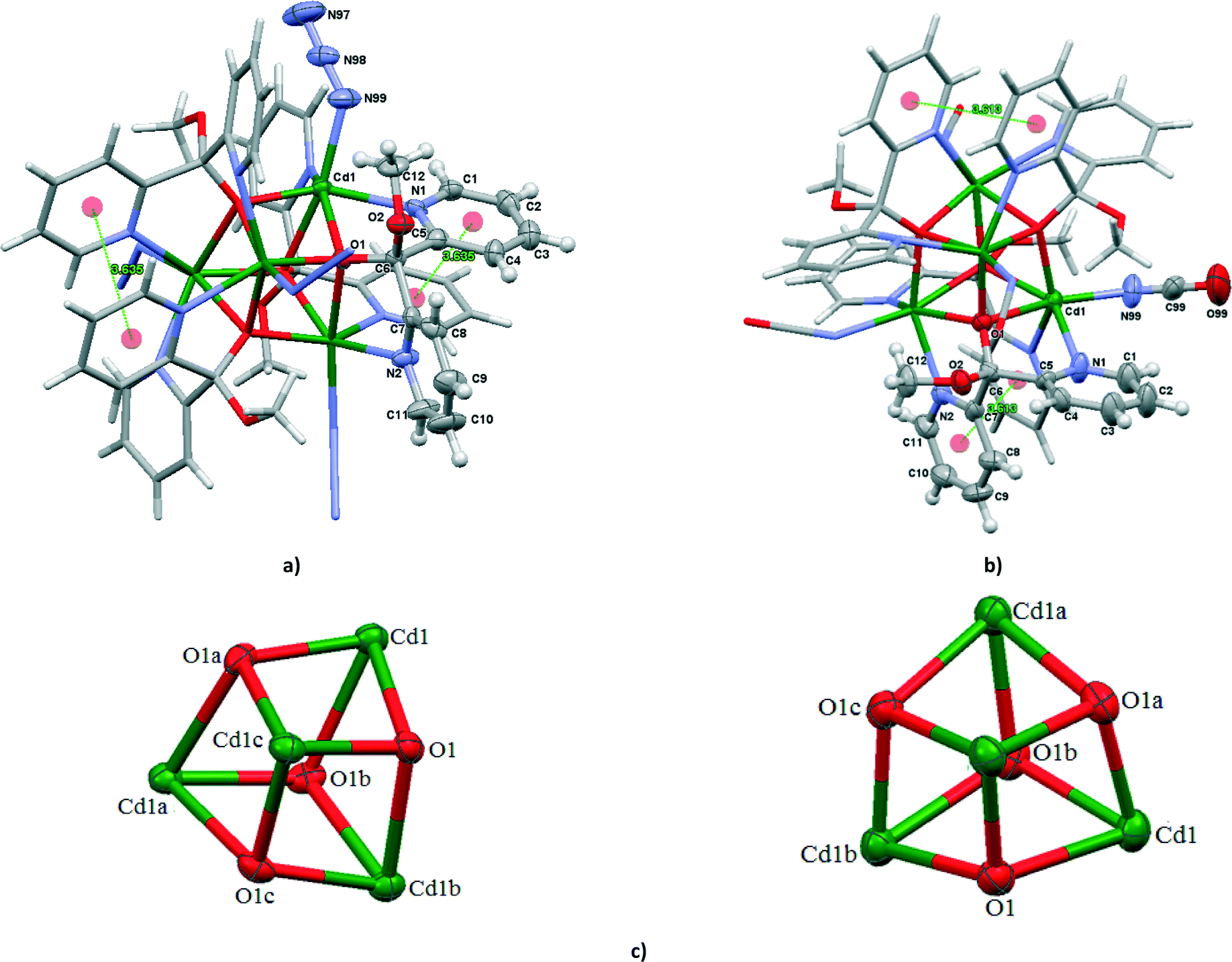 | ||
| Fig. 3 Molecular structures of [Cd4(N3)4{(py)2C(OCH3)(O)}4] (a) and [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (b). Displacement ellipsoids are drawn at 50% probability; (c) cubane unit schemes of [Cd4(μ3-O)4]4+ core in [Cd4(N3)4{(py)2C(OCH3)(O)}4] (symmetry code (a) = −3/4 − y, −1/4 + x, 1/4 − z; (b) = −1 − x, 1/2 − y, z, (c) = 1/4 + y, 3/4 − x, 1/4 − z) and [Cd4(NCO)4{(py)2C(OCH3)(O)}4] (symmetry code (a) 3/4 − y, −1/4 + x, 5/4 − z; (b) = 1 − x, 1/2 − y, z, −z + 5/4; (c) = 1/4 + y, 3/4 − x, 5/4 − z). | ||
Structures of [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5) and [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7)
The structures of 5 and 7 consist of the centrosymmetric tetranuclear (binuclear of binuclear) cadmium(II) compounds [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (5) and [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (7) showing double open cubane-like structures with two missing vertices (Fig. 4). The dicubane-like cores in 5 and 7 are stabilized by hydrogen bond O(4)–H(4A)⋯O(1)i [D⋯A distance = 2.717(3) Å and D–H⋯A angle = 171.0°; (i): −x, 1 − y, 1 − z in 5 and D⋯A distance = 2.738(7) Å and D–H⋯A angle = 171.0°; (i): 1 − x, 1 − y, −z in 7].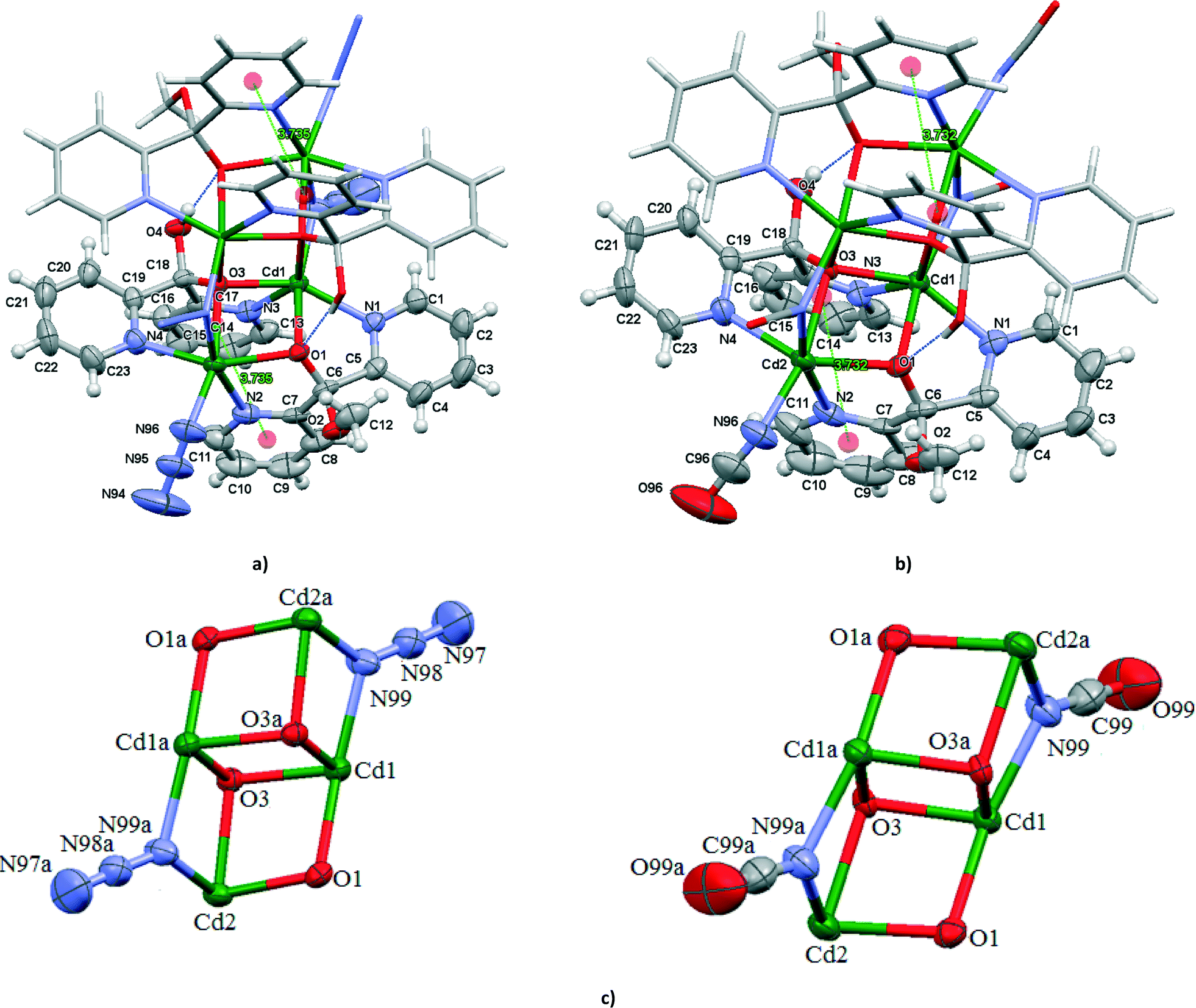 | ||
| Fig. 4 Molecular structures of [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (a) and [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2−] (b). Displacement ellipsoids are drawn at 50% probability; (e) dicubane unit schemes in [Cd4(N3)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (symmetry code (a) = −x, 1 − y, 1 − z) and [Cd4(NCO)4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2−] (symmetry code (a) = 1 − x, 1 − y, −z). | ||
The crystallographically related Cd(1) and Cd(1a) atoms are doubly bridged through the deprotonated oxygen atoms O(3) and O(3a) of the monoanion (py)2C(OH)O−. The atoms O(3) and O(3a) occupy two other vertices and act as triple bridges also bound to the Cd(2) and Cd(2a) atoms, respectively. In addition, the Cd(1) atoms are bridged to the Cd(2) through the deprotonated oxygen atom O(1) of the monoanion (py)2C(OCH3)O−, and to Cd(2a) through the nitrogen atom N(99) of the end-to-on N3− ion in 5 or end-to-on NCO− in 7. The monoanions (py)2C(OH)O− and (py)2C(OCH3)O− coordinate to the metal centers in an asymmetric way. The local environment around the Cd centers can best be described as a highly distorted octahedron. The Cd(1) atom is coordinated to two μ3-oxygen atoms of the monoanions (py)2C(OH)O−, one μ2-oxygen of (py)2C(OCH3)O−, two pyridine nitrogen atoms and nitrogen donor of μ1,1-N3− (in 5) or μ1,1-NCO− (in 7) bridge. The coordination sphere of the Cd(2) atom is composed of the μ3-oxygen atom of the monoanion (py)2C(OH)O−, μ2-oxygen atom of (py)2C(OCH3)O−, two pyridine nitrogen atoms and two nitrogen donors of the terminal and bridging ions, N3− in 5 and NCO− in 7. The angles Cd–Nazide–Cd (101.37(13)°) and Cd–NNCO–Cd (100.0(3)°) are similar to those reported in the literature for [Cd4(dafone)4Cl2(μ1,1-N3)4(μ1,1,1-N3)2] (93.49(18), 107.3(2) and 107.8(2)°)23 and [Cd(tp)(NCO)2]n (99.33(9) and 87.14(8)°).24 The pyridyl rings are involved in π–π interactions; 3.735(3) Å for Cg(18)ii((N(2)–C(7)–C(11))⋯Cg(19)((N(3)–C(13)–C(17)) centroids for 5 and 3.732(6) Å for Cg(18)ii((N(2)–C(7)–C(11))⋯Cg(19)((N(3)–C(13)–C(17)) centroids for 6 (ii) = x, y, z].
Structures of [Cd4(N3)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (8) and [Cd4(NCO)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (9a and 9b)
Like 1 and 7, 8, 9a and 9b are centrosymmetric tetranuclear (binuclear of binuclear) double open cubane-like compounds. In contrast to 1 and 7, however, they possess two nitrate groups instead of terminal azide ions (Fig. 5). Interestingly, the compound [Cd4{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2(NCO)2(NO3)2] was isolated in two polymorphic forms, monoclinic (9a), isostructural to 8, and orthorhombic (9b).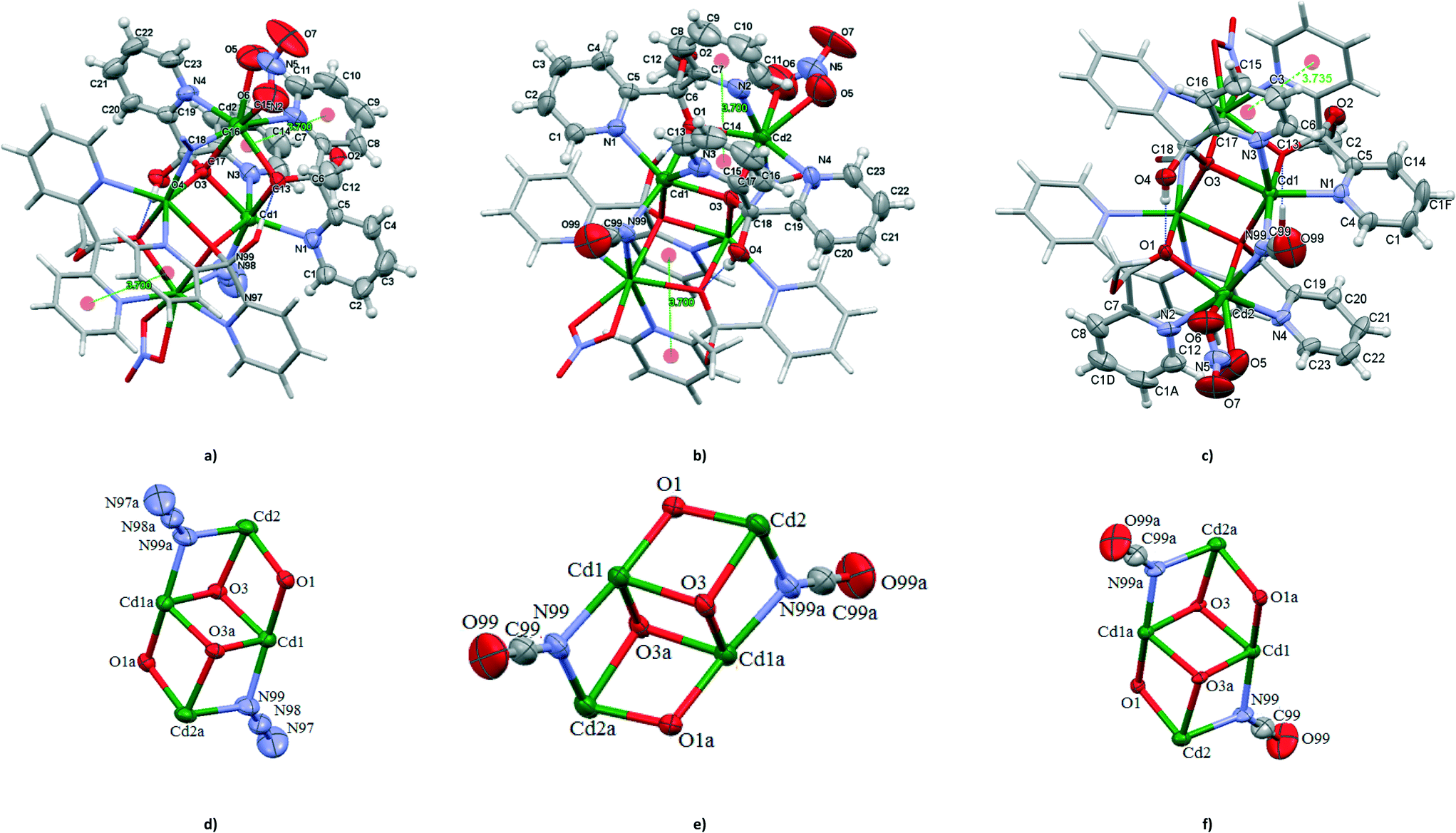 | ||
| Fig. 5 Molecular structures of [Cd4(N3)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (a) and [Cd4(NCO)2(NO3)2{(py)2C(OH)(O)}2{(py)2C(OCH3)(O)}2] (b and c). Displacement ellipsoids are drawn at 50% probability; dicubane unit schemes in 8 (d; symmetry code (a) = 1 − x, −y, 1 − z), in 9a (e; symmetry code (a) = −x, 1 − y, 1 − z) and 9b (f; symmetry code (a) = 1 − x, 1 − y, 1 − z). | ||
The Cd(1) atom of 8 and 9 is coordinated to two μ3-oxygen atoms of the monoanions (py)2C(OH)O−, one μ2-oxygen of (py)2C(OCH3)O−, two pyridine nitrogen atoms and nitrogen donor of μ1,1-N3− (in 5) or μ1,1-NCO− (in 7) bridge. The coordination environments of Cd(2) is formed by two N pyridine donors belonging to (py)2C(OCH3)O− and (py)2C(OCH3)O− ligands, nitrogen atom of the bridging pseudohalide ion (N3− in 8 and NCO− in 9), μ3-oxygen atom of the monoanion (py)2C(OH)O−, μ2-oxygen of (py)2C(OCH3)O− and two oxygen atoms of the nitrate group acting as chelating bidentate ligand. The geometrical distortion of the Cd(2) ions in 8 and 9 from ideal possible seven-vertex polyhedra (pentagonal bipyramid (PBPY), capped octahedron (COC) and capped trigonal prism (CTP)) have been calculated using the SHAPE program25 based on the Continuous Shape Measures (CShM) concept. Mathematically, the CShM of the original structure (Q) is a normalized root-mean-square deviation of the referenced structure with the desired symmetry (P) and it is expressed by eqn (1):
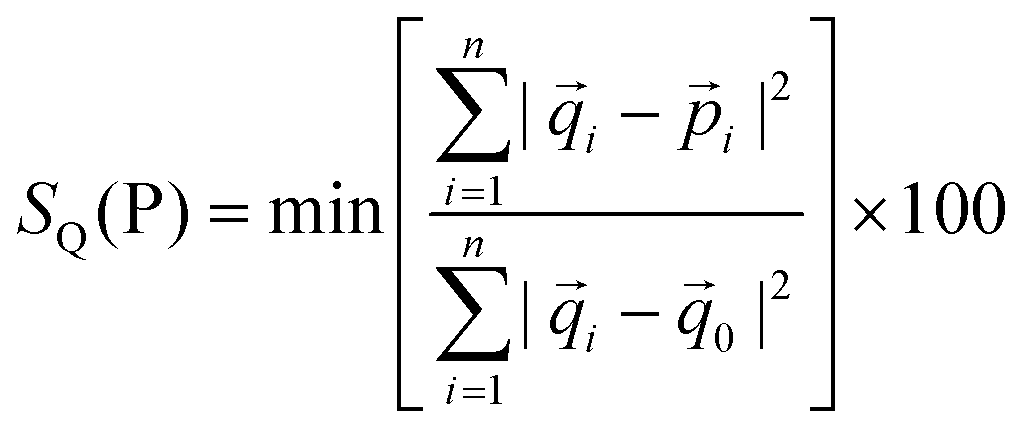 | (1) |
The calculated distances to the capped octahedron (5.043 for 8, 5.201 for 9a and 5.092 for 9b), capped trigonal prism (4.581 for 8, 4.904 for 9a and 4.211 for 9b) and PBPY (4.047 for 8, 3.995 for 9a and 4.074 for 9b) indicate severe distortions of the geometry of the Cd(2) ions in 8, 9a and 9b from all ideal seven-vertex polyhedra.
The bond lengths between the cadmium and oxygen atoms of the nitrate (2.427(5) and 2.491(6) Å in 8, 2.435(6) and 2.491(6) Å in 9a and 2.435(8) and 2.500(7) Å in 9b) are similar to the average value of 2.454(8) Å calculated for 55 coordination compounds incorporating the bidentate nitrate group and tridentate N-donor ligand; the mean value of the bond distance between the cadmium atom and the oxygen atom of the monodentate nitrate group calculated for 40 coordination compounds containing tridentate N-donor ligand is 2.401(1) Å.6 The bidentate fashion of nitrate ion in 8, 9a and 9b is also evidenced by the criteria introduced by Kleywegt, et al. (see Scheme 5).27
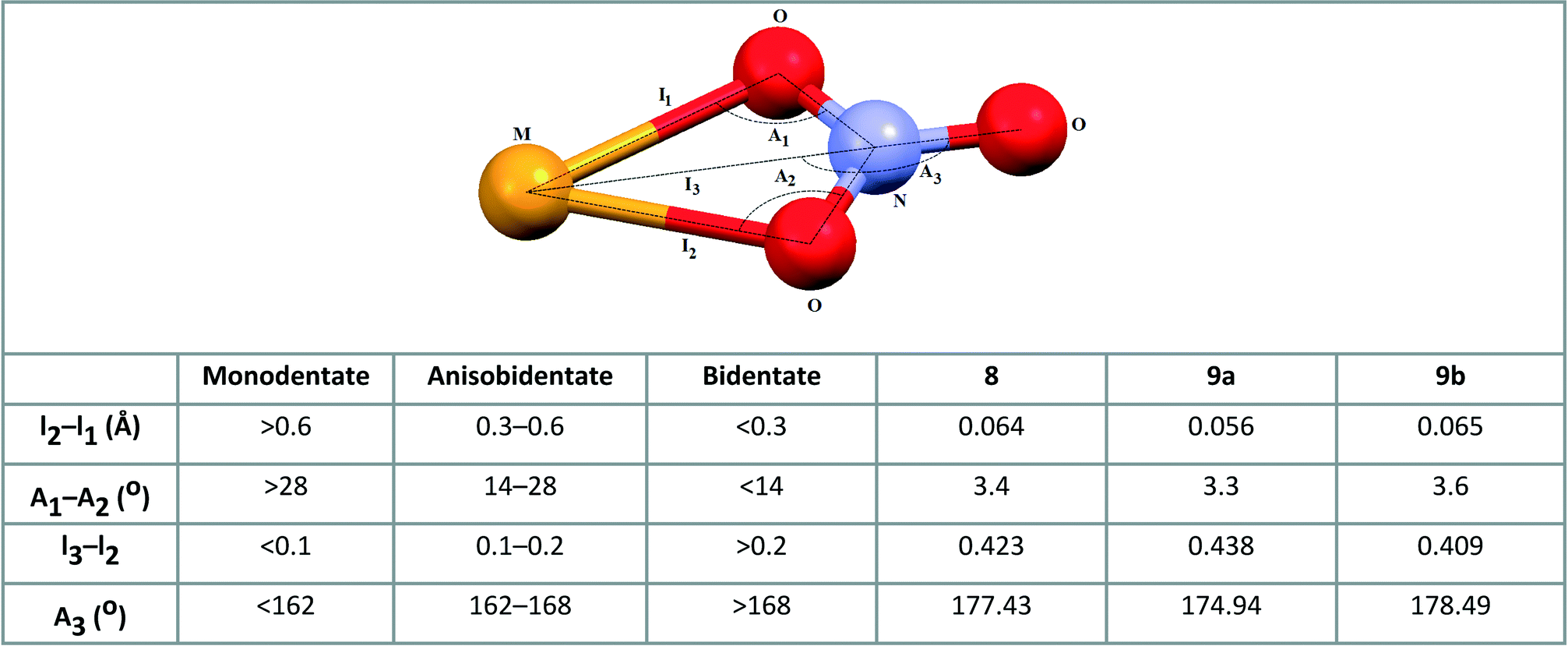 | ||
| Scheme 5 Parameters used for determining nitrate coordination mode27 and appropriate values for coordinated nitrate group in 8, 9a and 9b. | ||
Similar to 1 and 7, the dicubane-like core of 8 and 9 is stabilized by hydrogen bond O(4)–H(4A)⋯O(1)i [D⋯A distance = 2.712(4) Å and D–H⋯A angle = 177.0°; (i): 1 − x, −y, 1 − z in 8 and D⋯A distance = 2.707(7) Å and D–H⋯A angle = 171.0°; (i): −x, 1 − y, 1 − z in 9a and D⋯A distance = 2.705(6) Å and D–H⋯A angle = 177.0° in 9b]. Also, the structural parameters of the double open cubane-moiety of 8 and 9 correlate well with the values for 1 and 7.
The pyridyl rings are involved in π–π interactions; 3.700(4) Å for Cg(5)ii((N(2)–C(7)–C(11))⋯Cg(6)((N(3)–C(13)–C(17)) centroids for 8 and 3.700(5) Å for Cg(5)ii((N(2)–C(7)–C(11))⋯Cg(6)((N(3)–C(13)–C(17)) centroids for 9a, and 3.735(5) Å for Cg(6)iii((N(2)–C(7)–C(8)–C(10)–C(1a)–C(12))⋯Cg(7)((N(3)–C(13)–C(3)–C(15)–C(16)–C(17)) centroids for 9b, and (ii) = x, y, z; (iii) = 1 − x, 1 − y, 1 − z.
Luminescence properties
The excitation and emission spectra of the Cd(II) compounds and free (py)2CO were recorded in MeCN solution and in the solid state at room temperature. A summary of the photophysical data is provided in Table 1. The normalized emission spectra of 1, 2, 4–9 and free (py)2CO in MeCN and the solid state are shown in Fig. 6 and 7, respectively. The nanosecond range of lifetime in MeCN solution and the solid state suggests the fluorescent nature of the emissions (Table 1).| Compound | Medium | λ ex [nm] | λ em [nm] | Stokes shifts [cm−1] | τ, ns, (%) | χ 2 |
|---|---|---|---|---|---|---|
| (py) 2 CO | Solid | Non-emissive | ||||
| MeCN | 300 | 407 | 8763 | 1.62 (65.51%), 3.19 (34.49%) | 0.988 | |
| 1 | Solid | 437 | 509 | 3237 | 1.75 (39.07%), 6.47 (60.93%) | 1.024 |
| MeCN | — | — | — | — | — | |
| 2 | Solid | Non-emissive | ||||
| MeCN | 328 | 453 | 8413 | 2.55 | 1.134 | |
| 4 | Solid | 368 | 438 | 4343 | 0.88 (52.10%), 4.37 (47.90%) | 1.012 |
| MeCN | 365 | 449 | 5125 | 2.00 (63.11%), 6.09 (36.99%) | 1.223 | |
| 5 | Solid | 365 | 436 | 4461 | 0.69 (52.8%), 3.74 (47.2%) | 0.913 |
| MeCN | 345 | 460 | 7247 | 2.91 | 1.049 | |
| 6 | Solid | 351 | 427 | 5071 | 1.03 (60, 33%), 6.01 (39.67%) | 1.180 |
| MeCN | 339 | 457 | 7617 | 2.95 (100%) | 0983 | |
| 7 | Solid | 351 | 420/536 | 4680 | 0.79 (38.31%), 2.76 (33.19%), 6.55 (28.5%) | 1.194 |
| MeCN | 345 | 458 | 7152 | 2.94 | 1.011 | |
| 8 | Solid | 351 | 411 | 4159 | 0.66 (71.65%), 3.79 (28.37%) | 1.174 |
| MeCN | 342 | 457 | 7358 | 2.91 | 1.043 | |
| 9a and 9b | Solid | 360 | 440 | 5051 | 0.64 (80.77%) | 1.021 |
| MeCN | 335 | 446 | 7429 | 2.95 (80.77%) | 0.983 | |
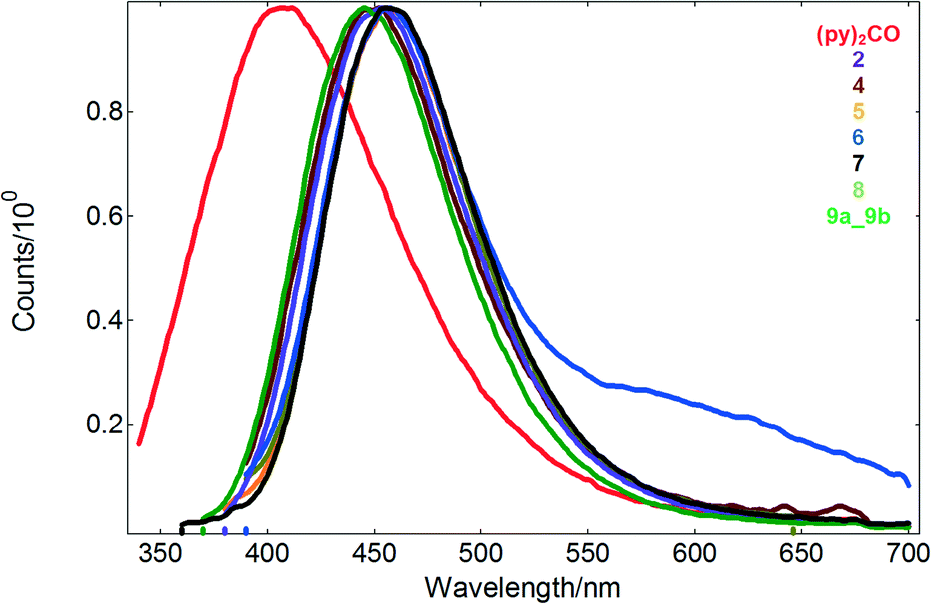 | ||
| Fig. 6 The emission spectra of the free ligand and Cd(II) compounds in acetonitrile solution (10−4 M). | ||
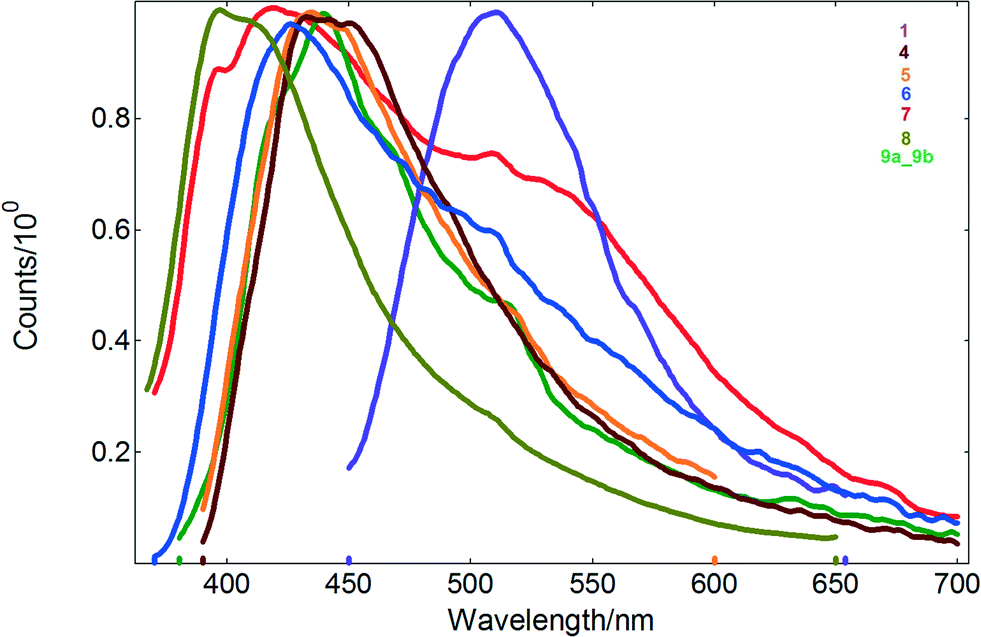 | ||
| Fig. 7 The solid state emission spectra of Cd(II) coordination compounds. | ||
Irradiation of 2 and 4–9 in solution with wavelengths of 328–365 nm gave rise to a structureless emission band with maxima in the ranges of 446–460 nm and Stokes shifts of 5125–8763 cm−1, respectively. Compared to the free ligand (λex = 300 nm, λex = 407 nm), the emission and excitation wavelengths of the coordination compounds are red-shifted (Table 1 and Fig. 6).
Taking into consideration that Cd(II) ions are difficult to oxidize or reduce due to their d10 configuration, however, it can be assumed that the emissions of 2 and 4–9 are neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature. It is highly probable that the emitting excited state of the Cd(II) compounds has an IL (intraligand) character. The red shift of the emission maximum with respect to the free ligand may be due to the following reasons: (i) the organic ligand may change their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels after coordination to metal centers; (ii) ligand to ligand charge transfer transitions may occur; and (iii) weak interactions may affect the energy transfer involved in the luminescence.
Except for 2, the reported Cd(II) compounds show also luminescence in the solid state, with emission maxima in the range of 411–536 nm and nanosecond lifetimes (Table 1 and Fig. 7). Noteworthy, di-2-pyridyl ketone is non-emissive in the solid state. For compound 7, the bimodal emission with maxima at 420 and 536 nm is observed, whereas excitation of the samples of 1, 4, 5, 6, 8 and 9 produces luminescence with one main peak at 509, 438, 436, 427, 411 and 440 nm, respectively.
Compared to the values reported for solution, the emission bands of 4, 5, 6, 8 and 9 appear blue-shifted, consistent with the rigidochromic effect. Furthermore, there is a marked difference in the fluorescence maximum wavelength of the helical structure of 1 with respect to that recorded for the tetranuclear compounds 4, 5, 6, 8 and 9 (Table 1). This effect is probably attributed to different coordination environments of the metal ions and conformational ligands as well as weak interactions in the network lattice which may affect the rigidity of the whole network and further the energy transfer involved in the luminescence.
The differences between the solid state and solution spectra of 9 (mixture of 9a and 9b) are analogous to the differences observed for other studied compounds (existing in the solid state as single polymorphic forms). Additionally the absorption/emission maxima visible in the solid state spectra of 9 do not show any effects suggesting that 9 is a physical mixture of 9a and 9b (which was proven by XRPD). This phenomenon originates from the character of the observed transitions. The emitting excited state of these compounds has an IL (intraligand) character. The electronic structure of the ligand is modified by participation of the ligand in the formation of the coordination bonds, but weak intermolecular interactions in the crystal lattice seem to have negligible effect on emissions of 9a and 9b.
Conclusions
The reactions of cadmium(II) nitrate with di-2-pyridyl ketone in the presence of pseudohalide (N3−, NCS− and NCO−) ions generated nine coordination compounds. The studies revealed that Cd2+ ions promote the conversion of the sp2 hybridized C atom in (py)2CO to sp3 (py)2C(OH)2 and (py)2C(OR)(OH). The inorganic anions not only balance the charges of the metal ion but also play a fundamental role in the formation of the final product and have a significant effect on its architecture. N3− and NCO− ions facilitate the formation of tetranuclear cadmium compounds which are cubane-like structures, whereas the reactions of Cd(NO3)2, (py)2CO and NH4SCN, which are dependent on the reaction variables, resulted in the formation of a helical 1D coordination polymer, a dimer as well as a cubane-like structure. The studies confirmed that the framework of dicubane-like cores in 5, 7, 8 and 9 is strongly reinforced by intramolecular O–H⋯N hydrogen bonds. Furthermore, the simultaneous presence of chelating nitrate and tridentate of organic ligands (py)2C(OH)(O)− and (py)2C(OCH3)(O)− appears to promote heptacoordination of Cd(II) ions in 8 and 9. Also, enhancement of fluorescence in the solid state compared to free ligand (py)2CO opens doors for new photochemical applications of these compounds, which are likely to be potential luminescent materials. One important advantage for the use of ligand-based emission in coordination compounds is that it may be tuned through variation of counteranions and/or the structure of the framework, as shown for the for examined compounds.Acknowledgements
Iwona Nawrot is deeply grateful for the financial support of the “DoktoRIS – Scholarship program for innovative Silesia” co- financed by the European Union under the European Social Fund.Notes and references
- R. R. Osborne and W. R. McWhinnie, J. Chem. Soc. A, 1967, 2075–2078 RSC.
- (a) A. C. Deveson, S. L. Heath, C. J. Harding and A. K. Powell, J. Chem. Soc., Dalton Trans., 1996, 3173–3178 RSC; (b) O. J. Parker, S. L. Aubol and G. L. Breneman, Polyhedron, 2000, 19, 623–626 CrossRef CAS.
- (a) Z. Serna, G. Barandika, R. Cortés, M. K. Urtiaga and M. I. Arriortua, Polyhedron, 1999, 18, 249–255 CrossRef CAS; (b) K. N. Crowder, S. J. Garcia, R. L. Burr, J. M. North, M. H. Wilson, B. L. Conley, P. E. Fanwick, P. S. White, K. D. Sienerth and Robert M. Graenger, Inorg. Chem., 2004, 43, 72–78 CrossRef CAS PubMed; (c) A. Khutia, P. J. S. Miguel and B. Lippert, Inorg. Chim. Acta, 2010, 363, 3048–3054 CrossRef CAS; (d) H. Sartzi, C. C. Stoumpos, M. Giouli, I. I. Verginadis, S. Ch. Karkabounas, L. Cunha-Silva, A. Escuer and S. P. Perlepes, Dalton Trans., 2012, 41, 11984–11988 RSC; (e) F. A. Mautner, M. S. El Fallah, O. Roubeau, S. Speed, S. J. Teat and R. Vicente, Eur. J. Inorg. Chem., 2013, 3483–3490 CrossRef CAS.
- (a) G. Yang, S.-L. Zheng, X.-M. Chen, H. Kay Lee, Z.-Y. Zhou and T. C. W. Mak, Inorg. Chim. Acta, 2000, 303, 86–93 CrossRef CAS; (b) E. Katsoulakou, N. Lalioti, C. P. Raptopoulou, A. Terzis, E. Manessi-Zoupa and S. P. Perlepes, Inorg. Chem. Commun., 2002, 5, 719–723 CrossRef CAS; (c) A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC; (d) Th. C. Stamatatos, C. G. Efthymiou, C. C. Stoumpos and S. P. Perlepes, Eur. J. Inorg. Chem., 2009, 3361–3391 CrossRef CAS; (e) J. C. Knight, A. J. Amoroso, P. G. Edwards, R. Prabaharan and N. Singh, Dalton Trans., 2010, 39, 8925–8936 RSC.
- (a) Z. E. Serna, M. K. Urtiaga, M. G. Barandika, R. Cortés, S. Martin, L. Lezama, M. I. Arriortua and T. Rojo, Inorg. Chem., 2001, 40, 4550–4555 CrossRef CAS PubMed; (b) G. S. Papaefstathiou, A. Escuer, F. A. Mautner, C. Raptopoulou, A. Terzis, S. P. Perlepes and R. Vicente, Eur. J. Inorg. Chem., 2005, 879–893 CrossRef CAS; (c) C. G. Efthymiou, C. P. Raptopoulou, A. Terzis, R. Boča, M. Korabic, J. Mrozinski, S. P. Perlepes and E. G. Bakalbassis, Eur. J. Inorg. Chem., 2006, 2236–2252 CrossRef CAS; (d) M.-Ch. Suen and J.-Ch. Wang, Inorg. Chem. Commun., 2006, 9, 478–481 CrossRef CAS; (e) S. K. Padhi and R. Sahu, Polyhedron, 2008, 27, 2662–2666 CrossRef CAS; (f) C. C. Stoumpos, I. A. Gass, C. J. Milios, E. Kefalloniti, C. P. Raptopoulou, A. Terzis, N. Lalioti, E. K. Brechin and S. P. Perlepes, Inorg. Chem. Commun., 2008, 11, 196–202 CrossRef CAS; (g) Z. Serna, N. De la Pinta, M. K. Urtiaga, L. Lezama, G. Madariaga, J. M. Clemente-Juan, E. Coronado and R. Cortés, Inorg. Chem., 2010, 49, 11541–11549 CrossRef CAS PubMed; (h) E. Katsoulakou, V. Bekiari, C. P. Raptopoulou, A. Terzis, E. Manessi-Zoupa, A. Powell and S. P. Perlepes, Inorg. Chem. Commun., 2011, 14, 1057–1060 CrossRef CAS; (i) C. J. Milios, P. Kyritsis, C. P. Raptopoulou, A. Terzis, R. Vicente, A. Escuer and S. P. Perlepes, Dalton Trans., 2005, 501–511 RSC; (j) N. Lalioti, C. P. Raptopoulou, A. Terzis, A. E. Aliev, I. P. Gerothanassis, E. Manessi-Zoupa and S. P. Perlepes, Angew. Chem., Int. Ed., 2001, 40, 3211–3214 CrossRef CAS; (k) A. K. Boudalis, Y. Sanakis, J. M. Clemente-Juan, B. Donnadieu, V. Nastopoulos, A. Mari, Y. Coppel, J.-P. Tuchagues and S. P. Perlepes, Chem. – Eur. J., 2008, 14, 2514–2526 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- Y.-M. Li, J.-L. Zhang and X.-W. Zhao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2475 CAS.
- H.-G. Zhu, G. Yang and X.-M. Chen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 969–970 Search PubMed.
- M. Esmhosseini, N. Safari and V. Amani, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m1434–m1435 CAS.
- CrysAlis RED, Oxford Diffraction Ltd., Version 1.171.35.11, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edfinfton, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th edn, John Wiley & Sons, Inc., New York, 1997 Search PubMed.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- D. Bose, Sk H. Rahaman, R. Ghosh, G. Mostafa, J. Ribas, Ch.-H. Hung and B. K. Ghosh, Polyhedron, 2006, 25, 645–653 CrossRef CAS.
- M. Cannas, G. Carta, A. Cristini and G. Marongiu, Inorg. Chem., 1977, 16, 228–230 CrossRef CAS.
- G. Mostafa, A. Mondal, I. R. Laskar, A. J. Welch and N. Ray Chaudhuri, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 146–148 Search PubMed.
- M. Iwaoka and N. Isozumi, Molecules, 2012, 17, 7266–7283 CrossRef CAS PubMed.
- Z.-L. You and H.-L. Zhu, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m397–m399 Search PubMed.
- D. L. Reger, T. D. Wright, M. D. Smith, A. L. Rheingold, S. Kassel, T. Concolino and B. Rhagitan, Polyhedron, 2002, 21, 1795–1807 CrossRef CAS.
- S. Ray, S. Konar, A. Jana, S. Jana, A. Patra, S. Chatterjee, J. A. Golen, A. L. Rheingold, S. S. Mandal and S. K. Kar, Polyhedron, 2012, 33, 82–89 CrossRef CAS.
- S. Banerjee, B. Wu, P.-G. Lassahn, C. Janiak and A. Ghosh, Inorg. Chim. Acta, 2005, 358, 535–544 CrossRef CAS.
- B. Machura, I. Nawrot and K. Michalik, Polyhedron, 2012, 31, 548–557 CrossRef CAS.
- A. B. Caballero, A. Rodríguez-Diéguez, E. Barea, M. Quirós and J. M. Salas, CrystEngComm, 2010, 12, 3038–3045 RSC.
- M. Llunell, D. Casanova, J. Cirera, M. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE program, version 1.1b, Barcelona, 2003 Search PubMed.
- (a) D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2006, 126, 1755–1763 CrossRef PubMed; (b) E. M. Zueva, E. R. Ryabikh and S. A. Borshch, Inorg. Chem., 2011, 50, 11143–11151 CrossRef CAS PubMed.
- G. J. Kleywegt, W. G. R. Wiesmeijer, G. J. Van Driel, W. L. Driessen, J. Reedijk and J. H. Noordik, J. Chem. Soc., Dalton Trans., 1985, 2177–2184 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculated XRPD spectra, IR spectra, DSC curves, electronic absorption spectra in the solid state and in acetonitrile solution. CCDC 1446466–1446474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00112b |
| This journal is © The Royal Society of Chemistry 2016 |