
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvothermal synthesis of coordination polymers at different temperatures and their luminescence studies†
Sanchari
Pal
,
Tapan K.
Pal
and
Parimal K.
Bharadwaj
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: pkb@iitk.ac.in
First published on 3rd February 2016
Abstract
A new linker, [1,1′:2′,1′′-terphenyl]-4′,5′-dimethoxy-4,4′′-dicarboxylic acid (H2L) has been used along with 4,4′-azobispyridine (azpy) as the co-linker to solvothermally synthesize six coordination polymers (CPs). These compounds are formulated as {[Zn(L)(azpy)0.5]·(0.5H2O)}n (1), {[Zn2(L)2(azpy)]·(DMF)·(1.5H2O)}n (2), {[Zn(L)(bphy)]·(DMF)}n (3), [Cd(L)(azpy)0.5(DMF)(H2O)]n (4), {[Co(L)(azpy)](2H2O)}n (5) and {[Co3(L)2(bphy)2(HCOO)2(H2O)2](6DMF)(6H2O)}n (6) (bphy = 1,2-bis(4-pyridyl)hydrazine). Interestingly, 1 and 2 which are synthesized at 90 and 120 °C, respectively, are found to be orientation isomers. When the solvothermal reactions are carried out at 140 °C, the azo-bond in the co-linker azpy is reduced to bphy as found in 3 and 6. All the complexes exhibit an sql topology. They are characterized by X-ray crystallography, elemental analysis, thermogravimetry, powder X-ray diffraction and infrared spectroscopy. Solid state photoluminescence studies show an intra-ligand π–π* transition in each case.
Introduction
The design and synthesis of coordination polymers (CPs) have acquired an explosive growth in recent years due to not only their fascinating architectures but also their potential applications in different contemporary fields.1–7 Synthesis of these materials demands designing of new linkers that can be combined with the coordination tendencies of the metal ions.8,9 Interestingly, temperature, solvent, concentration, pH of the medium and templating molecules (ions) can greatly influence the ultimate structure of these materials.10–13 Particularly, temperature can play an important role during synthesis, by influencing the rate of crystal growth and the final structure.14,15 Predictability of the resultant structure becomes very difficult when the linker can adopt more than one conformation.16,17 On the other hand, in situ generation of new ligands18 during the solvothermal synthesis as well as framework isomerism19–24 has added another dimension in the synthesis of new architectures.Herein, we report a new V-shaped linker, [1,1′:2′,1′′-terphenyl]-4′,5′-dimethoxy-4,4′′-dicarboxylic acid (H2L) which solvothermally forms six different coordination polymers when used with the co-linker 4,4′-azobispyridine (azpy) (Scheme 1). The co-linker has interesting structural features for studying optical and redox properties.25 Besides, the possibility of its in situ reduction26–31 to 1,2-bis(4-pyridyl)hydrazine (bphy) offers another route to synthesize new compounds. We report the successful synthesis of six new CPs and their photoluminescence properties at room temperature in solid state as well as in dispersed medium.
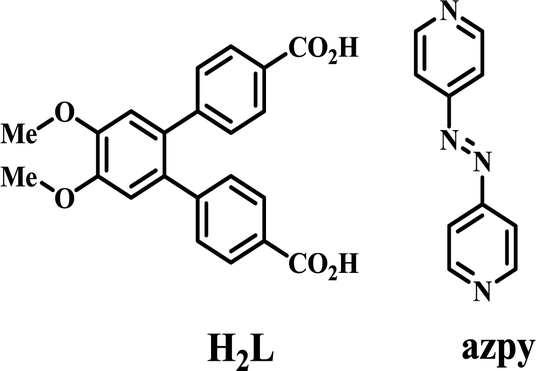 | ||
| Scheme 1 Schematic diagram of the linker H2L and co-linker azpy. | ||
Experimental section
Materials and measurements
Reagent grade 1,2-dimethoxybenzene, PdCl2, Zn(NO3)2·6H2O, Cd(NO3)2·4H2O, and Co(NO3)2·6H2O were obtained from Sigma-Aldrich and used as received. All solvents were procured from S. D. Fine Chemicals, India and they were purified following the established methods prior to use. Characterization of the linker and the co-linker were carried out by a number of spectroscopic techniques as detailed previously.32 Powder X-ray diffraction (PXRD) patterns of the compounds were recorded on a Bruker D8 Advance diffractometer equipped with nickel-filtered CuKα radiation. Thermogravimetric analyses (TGA) (5 °C min−1 heating rate under nitrogen atmosphere) were performed using a Mettler Toledo Star System. The solid-state emission spectra were recorded using a Jobin Yvon Horiba Fluorolog-3 spectrofluorimeter at room temperature (RT). The UV-vis spectra were recorded on a Shimadzu 2450 UV-vis spectrophotometer at RT. The steady-state emission spectra of the complexes dispersed in solvents were obtained using a Perkin-Elmer LS 50B luminescence spectrometer at RT with excitation and emission band-pass of 2.5 nm.X-ray crystallography
Single crystal X-ray diffraction data of 1–6 were collected at 100 K on a Bruker SMART APEX CCD diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The data reduction, structure solution and refinement were done as detailed earlier.32 In 6, the H atoms of the coordinated water molecules could not be located in the difference Fourier maps. In all the cases, the H atoms attached to C atoms were positioned geometrically and treated as riding atoms using SHELXL default parameters. Several DFIX commands were used for fixing a few bond distances in 2–6. Except in 3 and 4, the disordered solvent molecules could not be located in the successive difference Fourier maps and hence the PLATON33 squeeze method was used. The number of guest molecules present in the complexes was confirmed by the combination of thermogravimetric (TG) and elemental analyses. The crystal and refinement data for 1–6 are collected in Table S1† while selective bond distances and angles are given in Table S2.†Synthesis of the ligands
Details of the synthetic procedure for the ligand H2L and its characterization are given in the ESI.† The co-ligand azpy was synthesized following a literature procedure.34Synthesis of the complexes
Results and discussion
Once isolated, 1–6 are found to be stable in air and insoluble in water and common organic solvents. The IR spectra (Fig. S7–S12†) of all the complexes show strong absorption bands in the range of 1400–1600 cm−1 attributable to coordinated carboxylate groups.35 Broad bands in the region of 3230–3560 cm−1 indicate the presence of lattice and coordinated water molecules.36 Sharp peaks between 1640 and 1690 cm−1 are indicative of the presence of DMF molecules.37 The peak in the range of 1609–1632 cm−1 corresponds to the presence of –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– in the framework.38
N– in the framework.38
Complexes 1 and 2 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†). Both structures contain paddle-wheel Zn2(COO)4 clusters as the secondary building unit (SBU) where each Zn2+ ion adopts a distorted square-pyramidal coordination. The equatorial positions are occupied by the carboxylate O atoms from four different L−2 (hereafter, L) linkers and the apical position is occupied by a N atom of azpy co-linker (Fig. 1). All the Zn–O and Zn–N bond distances are comparable to the values in the existing literature.39
(Table S1†). Both structures contain paddle-wheel Zn2(COO)4 clusters as the secondary building unit (SBU) where each Zn2+ ion adopts a distorted square-pyramidal coordination. The equatorial positions are occupied by the carboxylate O atoms from four different L−2 (hereafter, L) linkers and the apical position is occupied by a N atom of azpy co-linker (Fig. 1). All the Zn–O and Zn–N bond distances are comparable to the values in the existing literature.39
 | ||
| Fig. 1 Metal–linker connectivity leads to isomeric complexes (a) 1 and (b) 2 as viewed along the a-axis (H atoms are omitted for clarity). | ||
The binding mode of the carboxylate groups of L and the N atom of azpy to the metal centre (μ4:η1:η1:η1:η1 and μ2:η1:η1, respectively) is the same for both 1 and 2 (Fig. S13a, b and g†). This connectivity pattern leads to two-fold interpenetrated 2D layers in each case (Fig. 2). Topological simplification with the Topos software40 shows an sql topology (4-c uninodal net) with point symbol {44·62} (Fig. S14 and S15†).
 | ||
| Fig. 2 Two-fold interpenetrated 2D layers of (a) 1 and (b) 2 viewed along the c-axis. | ||
Although both 1 and 2 exhibit the same binding mode of the linker and the co-linker, careful examination of the structures reveal that they constitute a pair of orientation isomers (Fig. 1). In 1, a number of C–H⋯π interactions (Table S3†) exist among the H atoms of the –OCH3 group of L of one 2D layer with the aromatic π orbitals (ring a or c, Fig. 1) of another layer. These non-bonding interactions are present in 2 as well. In 1, the aromatic ring of the azpy co-linker from the nearby layer is also involved in the C–H⋯π interactions with the –OCH3 group. However, this is absent in 2 (Fig. 3). Additionally, the interpenetrated layers in 2 show C–H⋯π interactions between the middle ring (ring b) of L and the –OCH3 group (Fig. 4), which is absent in 1. All these interactions (Fig. S16c and d†) are responsible for the formation of the orientation isomers.
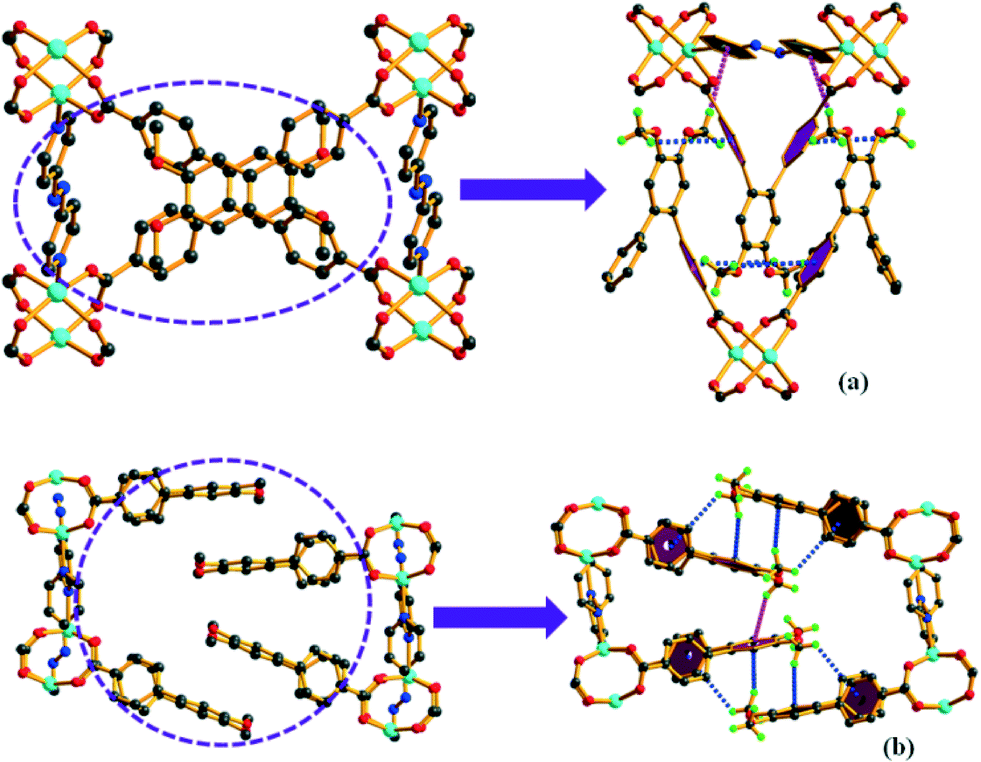 | ||
| Fig. 3 The perspective view of the C–H⋯π interactions in complex 1 (a) and 2 (b). | ||
 | ||
| Fig. 4 The C–H⋯π interactions (pink dotted lines) between two interpenetrated layers present in 2 (b) is absent in 1 (a). | ||
Complex 3 is formed under a solvothermal reaction temperature of 140 °C where the –NN– group in the co-linker azpy undergoes in situ reduction to –NH–NH– (bphy). It crystallizes in the orthorhombic space group Pbca (Table S1†) with the asymmetric unit consisting one Zn(II) ion, one L linker, one bphy co-linker and one DMF molecule in the cavity (Fig. S17a†). The metal ion assumes a distorted tetrahedral coordination geometry (Fig. 5a) with ligation by two pyridine N atoms and two monodentate carboxylate O atoms.
 | ||
| Fig. 5 (a) The perspective view of the coordination environment around the Zn(II) center, (b) the dotted lines represent the C–H⋯π and hydrogen bonding interactions. | ||
The C–N–N–C torsion angle in 3 is 106.22(65)° which indicates its deviation from the planar configuration of –C–NN–C– in azpy (nearly 180°) (Table S4†). The mode of binding of carboxylates and bphy ligands are: μ2:η1:η0:η1:η0 and μ2:η1:η1, respectively (Fig. S13c and h†). In 3, the bphy unit propagates along the b-axis and forms a 1D parallel chain that is connected by L leading to a 2D structure (Fig. S17b and c†). A number of non-covalent interactions (Fig. 5b) between these 2D layers lead to an overall 3D architecture. Topological simplification shows an sql topology (Fig. S18†).
Complex 4 crystallizes in the triclinic space group P (Table S1†). The asymmetric unit consists of one Cd(II) ion, one L unit, half of an azpy ligand, one coordinated water and DMF molecule (Fig. S19a†). The Cd2+ ion is hepta-coordinated with ligation by four O atoms of two bidentate carboxylates, one N atom of azpy and two O atoms of water and DMF molecules (Fig. S19b†). All Cd–O and Cd–N bond distances (Table S2†) are comparable to those reported earlier.32
The carboxylate and the azpy ligands bind to the metal ion in μ2:η1:η1:η1:η1 and μ2:η1:η1 modes, respectively (Fig. S13d and g†). Each L binds to a Cd(II) ion in a bidentate fashion at either end. Each metal ion is further connected to the N atoms of azpy to form a 1D structure (Fig. 6). These 1D chains are interconnected by strong H-bonding interactions between the coordinated water molecule and carboxylates besides and C–H⋯π interactions between the H atom of –OCH3 group and the π orbitals of the middle phenyl ring (ring b) of the nearest L ligand to form an overall 3D structure (Fig. S19†).
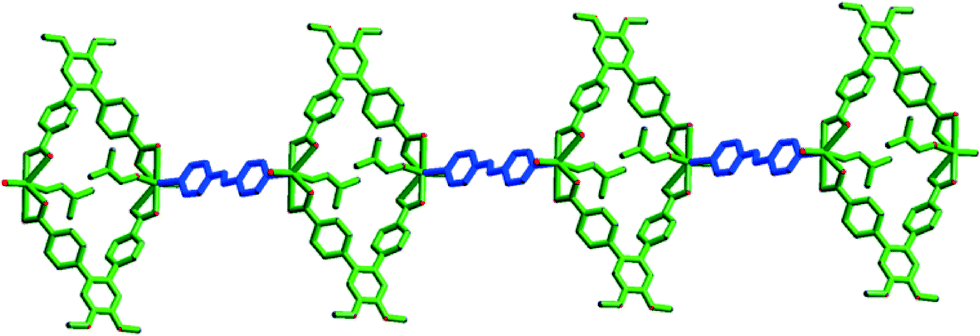 | ||
| Fig. 6 One dimensional chain of 4. | ||
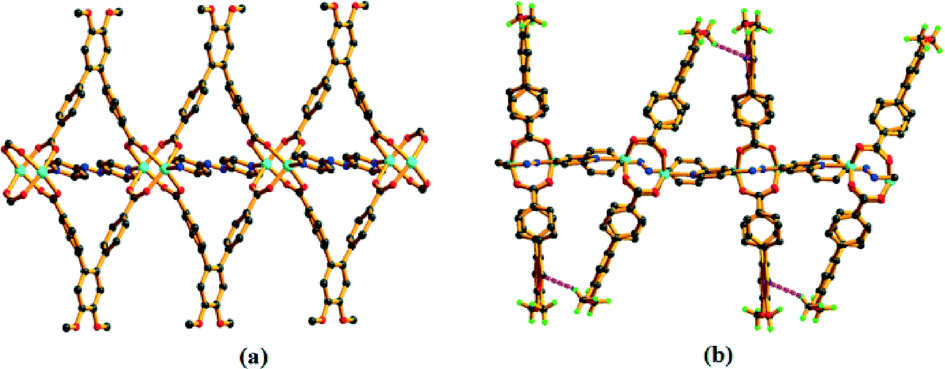
Complex 5 crystallizes in the monoclinic space group C2/m (Table S1†). The asymmetric unit comprises one L linker (half occupancy), one Co(II) ion (half occupancy) and one azpy co-linker (half occupancy) (Fig. S20a†). The structure contains paddle-wheel Co2(COO)4 clusters as the SBU, where the Co⋯Co distance is approximately 2.713(1) Å. Each Co(II) ion adopts a distorted square-pyramidal coordination geometry with equatorial ligation by carboxylate O atoms from four different L units and the axial site is occupied by one N atom from an azpy molecule (Fig. S20b†). The binding pattern of the carboxylate group of L and azpy to the metal centre are: μ4:η1:η1:η1:η1 and μ2:η1:η1, respectively (Fig. S13e and g†) and that forms a 2D layer structure (Fig. S20c†). These layers are further involved in the C–H⋯π interactions (Table S3†) between the H atom of the –OCH3 group and the π-orbitals of the phenyl rings of the azpy co-linker to form an overall 3D architecture (Fig. 7). All the Co–O and Co–N bond distances are comparable to those reported earlier (Table S2†).41 Complex 5 also exhibits an sql topology (Fig. S21†).
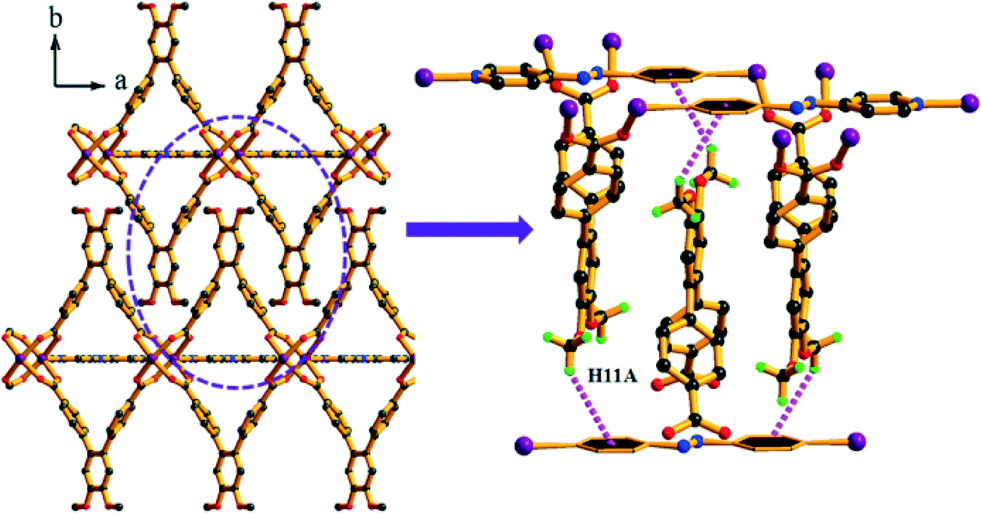 | ||
| Fig. 7 The presence of C–H⋯π interaction of 5 in crystallographic c direction. | ||
Compound 6 crystallizes in the monoclinic space group P21/c (Table S1†). The asymmetric unit contains two crystallographically independent Co(II) ions (Co1 half and Co2 full occupancy), one L and one bphy ligand, one coordinated formate and one coordinated water (Fig. S22†). In situ reduction of the azpy co-linker to bphy and hydrolysis of DMF to formate take place as the solvothermal reaction temperature is raised to 140 °C.42
As shown in Fig. 8, the structure contains a centrosymmetric [CoII3]6+ trimeric core where each metal is hexacoordinated. The middle Co(II) ion is ligated by four O atoms from four different L ligands and two O atoms from two bridging formate anions. Each terminal metal ion is coordinated by two O atoms from two different L ligands, two N atoms from two bphy ligands, one O atom from a water molecule and one O atom from one bridging formate anion. The reduction of the azo group has been confirmed by the N–N bond length and the –C–N–N–C– torsion angle (Table S4†). The binding modes of carboxylate and bphy ligands towards the metal ions are: μ4:η1:η1:η1:η1 and μ2:η1:η1, respectively (Fig. S13f and h†). These connectivity patterns lead to the formation of a 2D layer structure (Fig. 9) with an sql topology (Fig. S23†).
 | ||
| Fig. 8 Coordination environment around the Co(II) trimeric unit. | ||
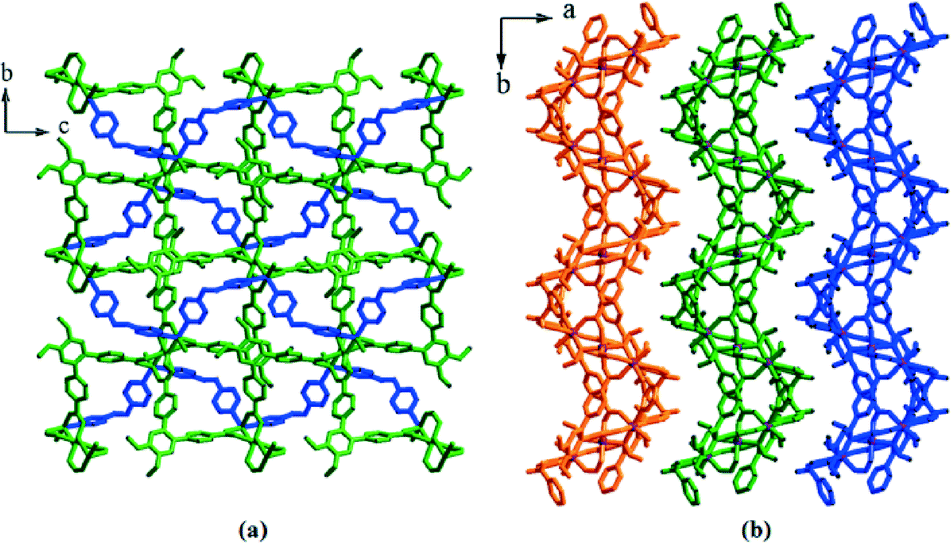 | ||
| Fig. 9 (a) View of a 2D layer in 6 as viewed along the a axis and (b) an overall structure of 6 as viewed along the c axis. | ||
PXRD and thermal stability
The phase purity of 1–6 can be confirmed by using powder X-ray diffraction (PXRD) patterns, which are in excellent agreement with the corresponding simulated patterns (Fig. S24–S29†).The TG curve of 1 shows a gradual weight loss of 1.48% (calcd. 1.65%) due to the removal of lattice water molecules in the temperature range of 30–100 °C. The framework is stable at least up to 385 °C (Fig. S30†). The TG curve of 2 exhibits a weight-loss of 2.1% till 110 °C attributable to the loss of lattice water molecules (calcd. 2.3%). Further heating leads to a rapid weight-loss of 6% in the range of 115–170 °C due to the loss of lattice DMF molecules (calcd. 6.26%). Decomposition of the compound is achieved beyond 380 °C (Fig. S31†). Complex 3 shows a weight loss of 10.30% corresponding to the loss of cavity DMF molecules (calcd. 10.40%) within 160 to 240 °C and after 355 °C it starts to decompose (Fig. S32†). Complex 4 releases its coordinated water (obsd. 2.7%; calcd. 2.6%) and DMF molecule (obsd. 10.5%; calcd. 11.16%) in the range of 95 to 105 °C and 175 to 205 °C, respectively and decomposition occurs after 360 °C (Fig. S33†). The TG curve of 5 displays gradual removal of its lattice water molecules (obsd. 5.6%; calcd. 5.48%) up to 130 °C and it decomposes after 380 °C (Fig. S34†). Complex 6 releases the lattice and coordinated water molecules in the range of 70 to 110 °C (obsd. 8.1%; calcd. 7.29%) and free DMF molecules up to 180 °C (obsd. 24.2%; calcd. 22.11%). Decomposition of this compound is achieved beyond 390 °C (Fig. S35†).
Photoluminescence properties
Photoluminescence studies of coordination polymers with d10 metal ions and conjugated ligands consisting of nitrogen and carboxylate donors have been a subject of investigation due to their potential applications43,44 in non-linear optics, sensors, photocatalysis and so on. Therefore, solid state luminescence properties of complexes 1–4 along with 5 and 6, free H2L and azpy are studied at room temperature. As shown in Fig. 10, the metal-free H2L shows an emission maximum at 450 nm upon excitation at 315 nm. On the other hand, metal-free azpy gives an emission at 412 nm upon excitation at 310 nm. The nature of the emission profiles of all the complexes except 3 and 6 resembled free azpy without any change of the emission maximum. Therefore, the nature of this emission is attributable to the intra-ligand n–π* or π–π* transition of azpy. The complexes with d10 metal ions, emission through metal-to-ligand or ligand-to-metal charge transfer is less probable due to difficulty in oxidation or reduction.45,46 Complexes 1, 2, 4 and 5 show a similar emission behaviour with a decrease in emission intensity compared to that of the free ligand probably due to the quenching effect of the water molecules present in the frameworks.47,48 For complex 5, the presence of the paramagnetic Co(II) ion can be a probable reason for its very low emission intensity.49,50 | ||
| Fig. 10 Emission spectra of H2L, azpy and 1–6 in solid state at room temperature. | ||
When azpy is in situ reduced to bphy, the π-conjugation is diminished and the energy gap between excited and ground states of bphy become higher than that in azpy, giving emission maxima at shorter wavelengths51 (392 and 386 nm for 3 and 6, respectively, λex = 340 nm), which can be attributed to the intra-ligand transition of the bphy ligand. The luminescence enhancement in 3 can result from the increased rigidity of the ligand upon metal coordination which reduces nonradiative decay.52 For 6, the fall in intensity is due to Co(II) as well as the vibrational quenching of water present in the framework. The emission spectra of 1–4 dispersed in solvents of different polarity (DMF, ethanol and hexane) do not show any significant shift of the emission bands obtained in the solid-state (Fig. S36†).
Conclusions
Six metal organic frameworks have been synthesized by the solvothermal reaction of a rationally designed V-shaped linker H2L and co-linker azpy with different metal ions at different temperatures. Among them, 1 and 2 exhibit temperature-dependent framework orientational isomerism due to the presence of different C–H⋯π interactions between two adjacent 2D layers which is a rare observation. Employment of a higher temperature leads to the in situ reduction of azpy ligand to the flexible bphy to afford 3 and 6, respectively. The room temperature photoluminescence properties were studied in solid-state. The successful synthesis of these complexes (1–6) improves the idea of crystal engineering which can help us to design ligands for the synthesis of coordination polymers endowed with interesting properties.Acknowledgements
We gratefully acknowledge the financial support from the Department of Science and Technology, New Delhi, India (to P. K. B.) and SRF from the CSIR to S. P. and T. K. P.Notes and references
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. J. Kim, M. O'Keefe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- K. Biradha, A. Ramanan and J. J. Vittal, Cryst. Growth Des., 2009, 9, 2969–2970 CAS.
- A. Santra, I. Senkovska, S. Kaskel and P. K. Bharadwaj, Inorg. Chem., 2013, 52, 7358–7366 CrossRef CAS PubMed.
- C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181–5184 CrossRef CAS PubMed.
- A. Aijaz, T. Akita, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2013, 135, 16356–16359 CrossRef CAS PubMed.
- D. De, T. K. Pal, S. Neogi, S. Senthilkumar, D. Das, S. S. Gupta and P. K. Bharadwaj, Chem. – Eur. J., 2016 DOI:10.1002/chem.201504747.
- S. S. Nagarkar, S. M. Unni, A. Sharma, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2638–2642 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- J. L. Atwood, L. J. Barbour and A. Jerga, Angew. Chem., Int. Ed., 2004, 43, 2948–2950 CrossRef CAS PubMed.
- Z. Su, J. A. Fan, M. Chen, T. Okamura and W. Y. Sun, Cryst. Growth Des., 2011, 11, 1159–1169 CAS.
- C. N. Morrison, A. K. Powell and G. E. Kostakis, Cryst. Growth Des., 2011, 11, 3653–3662 CAS.
- Q. A. Zhang, J. Y. Zhang, Q. Y. Yu, M. Pan and C. Y. Su, Cryst. Growth Des., 2010, 10, 4076–4084 CAS.
- A. Santra and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 1476–1485 CAS.
- Y.-B. Dong, Y.-Y. Jiang, J. Li, J.-P. Ma, F.-L. Liu, B. Tang, R.-Q. Huang and S. R. Batten, J. Am. Chem. Soc., 2007, 129, 4520–4521 CrossRef CAS PubMed.
- S. Masaoka, D. Tanaka, Y. Nakanishi and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2530–2534 CrossRef CAS PubMed.
- P. Cui, J. Dou, D. Sun, F. Dai, S. Wang, D. Sun and Q. Wu, CrystEngComm, 2011, 13, 6968–6971 RSC.
- M. V. Narinho, M. I. Yoshida, K. J. Guedes, K. Krambrock, A. J. Bortoluzzi, M. Horner, F. C. Machado and W. M. Teles, Inorg. Chem., 2004, 43, 1539–1544 CrossRef PubMed.
- G. B. Li, J. M. Liu, Z. Q. Yu, W. Wang and C. Y. Su, Inorg. Chem., 2009, 48, 8659–8661 CrossRef CAS PubMed.
- T. A. Makal, A. A. Yakovenko and H.-C. Zhou, J. Phys. Chem. Lett., 2011, 2, 1682–1689 CrossRef CAS.
- X. N. Zhao, H. He, F. Dai, D. Sun and Y. Ke, Inorg. Chem., 2010, 49, 8650–8652 CrossRef CAS PubMed.
- D. Sun, S. Ma, J. M. Simmons, J.-R. Li, D. Yuan and H.-C. Zhou, Chem. Commun., 2010, 46, 1329–1331 RSC.
- L. Han, A. Qin, X.-Z. Yan, L.-P. Xu, J. Sun, L. Yu, H.-B. Chen and X. Zou, Cryst. Growth Des., 2013, 13, 1807–1811 CAS.
- S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed.
- T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2013, 49, 6197–6199 RSC.
- R. Reuter and H. A. Wegner, Chem. Commun., 2011, 47, 12267–12276 RSC.
- X.-M. Liu, L.-H. Xie, J.-B. Lin, R.-B. Lin, J.-P. Zhang and X.-M. Chen, Dalton Trans., 2011, 40, 8549–8554 RSC.
- Y.-L. Gai, F.-L. Jiang, K.-C. Xiong, L. Chen, D.-Q. Yuan, L.-J. Zhang, K. Zhou and M.-C. Hong, Cryst. Growth Des., 2012, 12, 2079–2088 CAS.
- X.-M. Liu, R.-B. Lin, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 5686–5692 CrossRef CAS PubMed.
- J.-S. Guo, M.-J. Zhang, C.-J. Zhang, X.-M. Jiang, G.-C. Guo and J.-S. Huang, Inorg. Chem. Commun., 2013, 37, 206–210 CrossRef CAS.
- J.-S. Guo, G. Xu, X.-M. Jiang, M.-J. Zhang, B.-W. Liu and G.-C. Guo, Inorg. Chem., 2014, 53, 4278–4280 CrossRef CAS PubMed.
- J.-S. Guo, G. Xu, S.-H. Wang, M.-S. Wang, M.-J. Zhang, G.-C. Guo and J.-S. Huang, Inorg. Chem. Commun., 2014, 45, 108–111 CrossRef CAS.
- T. K. Pal, R. Katoch, A. Garg and P. K. Bharadwaj, Cryst. Growth Des., 2015, 15, 4526–4535 CAS.
- A. L. Spek, PLATON, The University of Utrecht, Utrecht, The Netherlands, 1999 Search PubMed.
- O. Theilmann, W. Saak, D. Haase and R. Beckhaus, Organometallics, 2009, 28, 2799–2807 CrossRef CAS.
- K. Nakamoto, Infrared and raman spectra of inorganic and coordination compounds, Wiley and sons, New York, 5th edn, 1997 Search PubMed.
- D. Dobrzynska, L. B. Jerzykiewicz, J. Jezierska and M. Duczmal, Cryst. Growth Des., 2005, 5, 1945–1951 CAS.
- T. K. Pal, D. De, S. Neogi and P. K. Bharadwaj, Inorg. Chem. Front., 2015, 2, 395–402 RSC.
- B. Bhattacharya, D. Saha, D. K. Maity, R. Dey and D. Ghoshal, CrystEngComm, 2014, 16, 4783–4795 RSC.
- T. K. Pal, S. Neogi and P. K. Bharadwaj, Chem. – Eur. J., 2015, 21, 16083–16090 CrossRef CAS PubMed.
- The network topology was evaluated by the program “TOPOS-4.0”, see http://www.topos.ssu.samara.ru; V. A. Blatov, IUCr Comp. Comm. Newsl., 2006, 7, 4 Search PubMed.
- P. Lama, J. Mrozinski and P. K. Bharadwaj, Cryst. Growth Des., 2012, 12, 3158–3168 CAS.
- (a) L. Xie, S. Liu, B. Gao, C. Zhang, C. Sun, D. Li and Z. Su, Chem. Commun., 2005, 2402–2404 RSC; (b) J.-J. Shen, M.-X. Li, Z.-X. Wang, C.-Y. Duan, S.-R. Zhu and X. He, Cryst. Growth Des., 2014, 14, 2818–2830 CrossRef CAS.
- F. P. Doty, C. A. Bauer, A. J. Skulan, P. G. Grant and M. D. Allendorf, Adv. Mater., 2009, 21, 95–101 CrossRef CAS.
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137–12139 RSC.
- L. Wen, Y. Li, Z. Lu, J. Lin, C. Duan and Q. Meng, Cryst. Growth Des., 2006, 6, 530–537 CAS.
- L. P. Zhang, J. F. Ma, J. Yang, Y. Y. Pang and J. C. Ma, Inorg. Chem., 2010, 49, 1535–1550 CrossRef CAS PubMed.
- A. J. Lan, K. H. Li, H. H. Wu, L. Z. Kong, N. Nijem, D. H. Olson, T. J. Emge, Y. J. Chabal, D. C. Langreth, M. C. Hong and J. Li, Inorg. Chem., 2009, 48, 7165–7173 CrossRef CAS PubMed.
- F. F. Chen, Z. Q. Chen, Z. Q. Bian and C. H. Huang, Coord. Chem. Rev., 2010, 254, 991–1010 CrossRef CAS.
- H.-F. Zhu, J. Fan, T. Okamura, W.-Y. Sunand and N. Ueyama, Cryst. Growth Des., 2005, 5, 289–294 CAS.
- K. J. Franz, N. Singh, B. Spingler and S. J. Lippar, Inorg. Chem., 2000, 39, 4081–4092 CrossRef CAS PubMed.
- J. D. Luo, J. L. Hua, J. G. Qin, J. Q. Cheng, Y. C. Shen, Z. H. Lu, P. Wang and C. Ye, Chem. Commun., 2001, 171–172 RSC.
- S. L. Zheng, J. H. Yang, X. L. Yu, X. M. Chen and W. T. Wong, Inorg. Chem., 2004, 43, 830–838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the ligands and their 1H, 13C-NMR, mass spectra, X-ray crystallographic data in CIF format, table for selected bond distances and angles for complexes 1–6, complete data for IR, PXRD, TGA analysis, and additional crystal structures. CCDC 1437804 (1), 1437805 (2), 1437806 (3), 1437807 (4), 1437808 (5), 1437809 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02540k |
| This journal is © The Royal Society of Chemistry 2016 |