
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new nanocrystalline binary phase: synthesis and properties of cubic tin monoselenide†
Ran E.
Abutbul
ab,
Elad
Segev
b,
Shmuel
Samuha
ac,
Leila
Zeiri
bd,
Vladimir
Ezersky
b,
Guy
Makov
ab and
Yuval
Golan
*ab
aDepartment of Materials Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: ygolan@bgu.ac.il
bIlse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
cNRCN, P.O. Box 9001, Beer-Sheva 84190, Israel
dChemistry Department, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
First published on 15th February 2016
Abstract
A new nanometric cubic binary phase of the tin mono-selenide system, π-SnSe, was obtained as cube shaped nanoparticles. Its structure and atomic positions were adopted from previously reported π-SnS (P213, a0 = 11.7 Å). The proposed structure model of π-SnSe, with 64 atoms per unit cell, was refined against experimental X-ray diffraction using Rietveld method (a0 = 11.9702(9) Å; Rp = 1.65 Rwp = 2.11). The optical properties of this new cubic SnSe phase were characterized by Raman and optical absorption spectroscopies. The optical band gap was assessed to be indirect, with Eg = 1.28 eV (in the near infrared), compared to Eg = 0.9 eV (indirect) and 1.3 eV (direct) for the conventional orthorhombic phase of α-SnSe. Raman spectroscopy indicated significant phonon restraining, which is likely to be beneficial for thermoelectric applications. Since the new cubic phase belongs to a class of non-centrosymmetric crystals, interesting and potentially useful properties may arise. Density functional theory calculations have been applied in order to validate phase stability and evaluate the energy bandgap. These results, together with the recently discovered cubic phase of π-SnS, confirm the existence of a new class of nanoscale materials in the tin chalcogenide system.
Introduction
Considerable attention is being focused on narrow band gap materials because of their potential use in solar cells, optical communication and near-infrared (NIR) detectors.1,2 In particular, tin mono-sulfide (SnS) and tin mono-selenide (SnSe) which belong to the IV–VI semiconductor family have become attractive due to their promising opto-electronic properties, moderate toxicity, chemical stability and natural abundance.1 Conventional tin-chalcogenides adopt a layered orthorhombic structure (GeS type) with two smaller lattice constants and one much larger. Because of the resemblance in atomic positions, this structure could also be viewed as a distorted NaCl structure. Indeed, much effort has been directed toward improved syntheses of tin-chalcogenide nanomaterials.4–14In the case of SnS, surfactant assisted synthesis of nanoparticles and chemical bath deposition (CBD) usually resulted in elongated platelet nanoparticles and flake like morphology respectively which corresponded to the orthorhombic structure of SnS referred to as α-SnS.12–21 Yet, more recent publications reported on zinc blende (ZB) nanoparticles or thin films which formed under specific synthetic conditions.13,22–26 Transmission electron microscopy (TEM) of the materials was inconsistent with the lattice parameter expected for the ZB structure. In addition, density functional theory (DFT) calculations predicted this phase to be thermodynamically unstable.27 We have recently reported that the new phase of SnS in fact belongs to a new, simple cubic binary phase of SnS, which possesses an exceptionally large lattice parameter (a0 = 11.7 Å)28 and which we designated as π-SnS.28 The equivalent orthorhombic analogue of α-SnS, tin mono-selenide (α-SnSe), is an indirect band gap semiconducting material (0.9 eV) with promising potential use in thermoelectric applications.7,10,29,30 In fact, the highest figure of merit for thermoelectric material efficiency has been reported for α-SnSe at elevated temeperature.29 SnS and SnSe share much in common, chemically and structurally. Hence, it was natural to question if the selenide analogue of the π-SnS prototype also existed in the form of π-SnSe.
In this publication, we report on a new cubic binary phase of tin mono-selenide, π-SnSe with a correspondingly large lattice parameter of a0 = 11.9702(9) Å. The described synthetic protocol enables synthesis of pure samples of α-SnSe and π-SnSe upon demand, allowing for their characterization in the nanoscale as well as in the macroscale. The π-SnSe was characterized using an array of powerful techniques, including X-ray diffraction (XRD), Raman spectroscopy and optical absorption spectroscopy. The stability of the π-SnSe has been studied using DFT calculation along with evaluation of the material band gap.
Experimental
(a) Materials
Purified water (18.2 MΩ cm) was obtained from Millipore filtration system. Tin(II) chloride (SnCl2, 98%), oleylamine (OLA, >98%), oleic acid (OA, >90%), 1-dodecanthiol (DDT, 98%), selenourea (SU, >98%), were all purchased from Sigma-Aldrich and used without further purification (unless stated otherwise in the text). Methanol (99.8%) and chloroform (99.9%) were purchased from Bio-Lab, Israel and used without further purification.(b) Synthesis of π-SnSe nanoparticles
5 ml of OLA (15.53 mmol) held under vacuum overnight, 56.88 mg of SnCl2 (0.3 mmol) 100 μL of water (5.5 mmol) were placed in a 50 ml three necked flask together with a magnetic stirrer bar. The flask was transferred to a heating mantle, evacuated and flushed with argon 4 times and stirred at 50 °C until a milky white suspension appeared. Then, 0.5 ml of OA was injected into the flask, which was then evacuated and left under vacuum for 30 min. Simultaneously, 1 ml of OLA (3.1 mmol) and 18.45 mg of SU (0.15 mmol) were placed in an amber vial together with a magnetic stirring bar. The vial was evacuated and flushed with argon 4 times and left under vacuum for 30 min. Subsequently, the reaction flask was flushed with argon and the temperature was raised to 120 °C at a heating rate of 10 degrees per min. Once the reaction reached the destination temperature, the milky white colour disappeared to form a clear yellowish solution. The reaction was left for another 30 min while in the meantime the amber vial was transferred a bench-top sonicating bath to dissolve any residual aggregates of SU. The SU–OLA solution was injected to the reaction flask. The reaction turned reddish-brown within 10 seconds after injection. The incubation time was set to 5 min which after the heating mantle was removed. The reaction was allowed to cool naturally to 40 °C and 1 ml of DDA was injected. The colloidal suspension was then stirred for another 5 min and 5 ml of chloroform were added into the flask. The solution was then washed with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of chloroform:methanol and centrifuged at 2800 rpm for 5 min.
:10 ratio of chloroform:methanol and centrifuged at 2800 rpm for 5 min.
(c) Synthesis of α-SnSe nanoparticles
α-SnSe nanoparticles were synthesized similarly as the synthesis of the π-SnSe by keeping all reaction procedures and parameters identical, except for elevating the temperature of the reaction to 240 °C instead of 120 °C. After injecting the SU–OLA solution, the reaction solution instantly turned black.Results and discussion
The two syntheses described above resulted in two different morphologies, thick platelets (Fig. 1a) and aggregated cube-like nanoparticles (Fig. 1b, e and f). The nanoparticles with thick platelet morphology exhibited wider size and shape distributions. The corresponding XRD pattern presented in Fig. 2a match to α-SnSe (JCPDS no. 48-1224, a0 = 11.498 Å, b0 = 4.153 Å, c0 = 4.440 Å). The cube-like nanoparticles were subjected to EDS chemical analysis in TEM, and showed a Sn:Se ratio of 1:1 within the experimental error (5%). Selected area electron diffraction (SAED) taken from a large cluster of nanoparticles yielded a ring pattern which could not be indexed as α-SnSe. A line profile was extracted from the SAED pattern by radial integration of the electron diffraction ring intensities in order to obtain the precise position of the reflections (Fig. 1c and d).3 The measured dhkl-spacings acquired from the line plot differed substantially from the dhkl-spacing expected for α-SnSe. The XRD pattern acquired from the same sample corroborated these results (Fig. 2b).
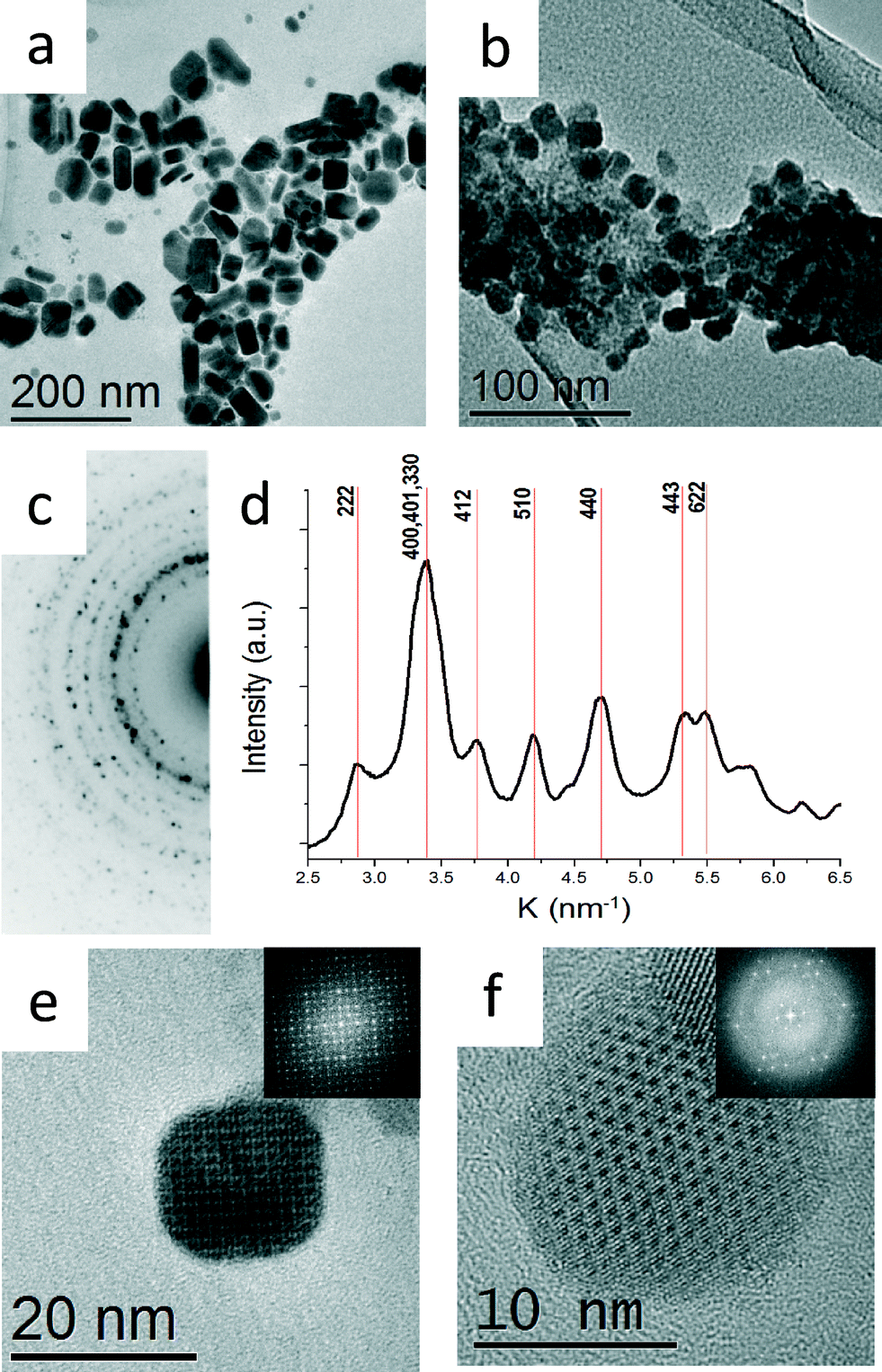 | ||
| Fig. 1 Bright field TEM micrographs showing (a) α-SnSe nanoparticles synthesized at 240 °C (b) π-SnSe nanoparticles synthesized at 120 °C. (c) SAED taken from b. (d) Intensity profile deduced from the diffraction pattern shown in c obtained by radial integration of the diffraction ring intensities.3 (e) HRTEM micrograph showing a cube-like nanoparticle taken along [001] orientation. Inset: FFT of the HRTEM image (f) HRTEM micrograph showing a cube-like nanoparticle taken along [111] orientation. Inset: FFT of the HRTEM image. Both SAED and FFTs confirmed the proposed cubic structure with a0 = 12.1 Å. | ||
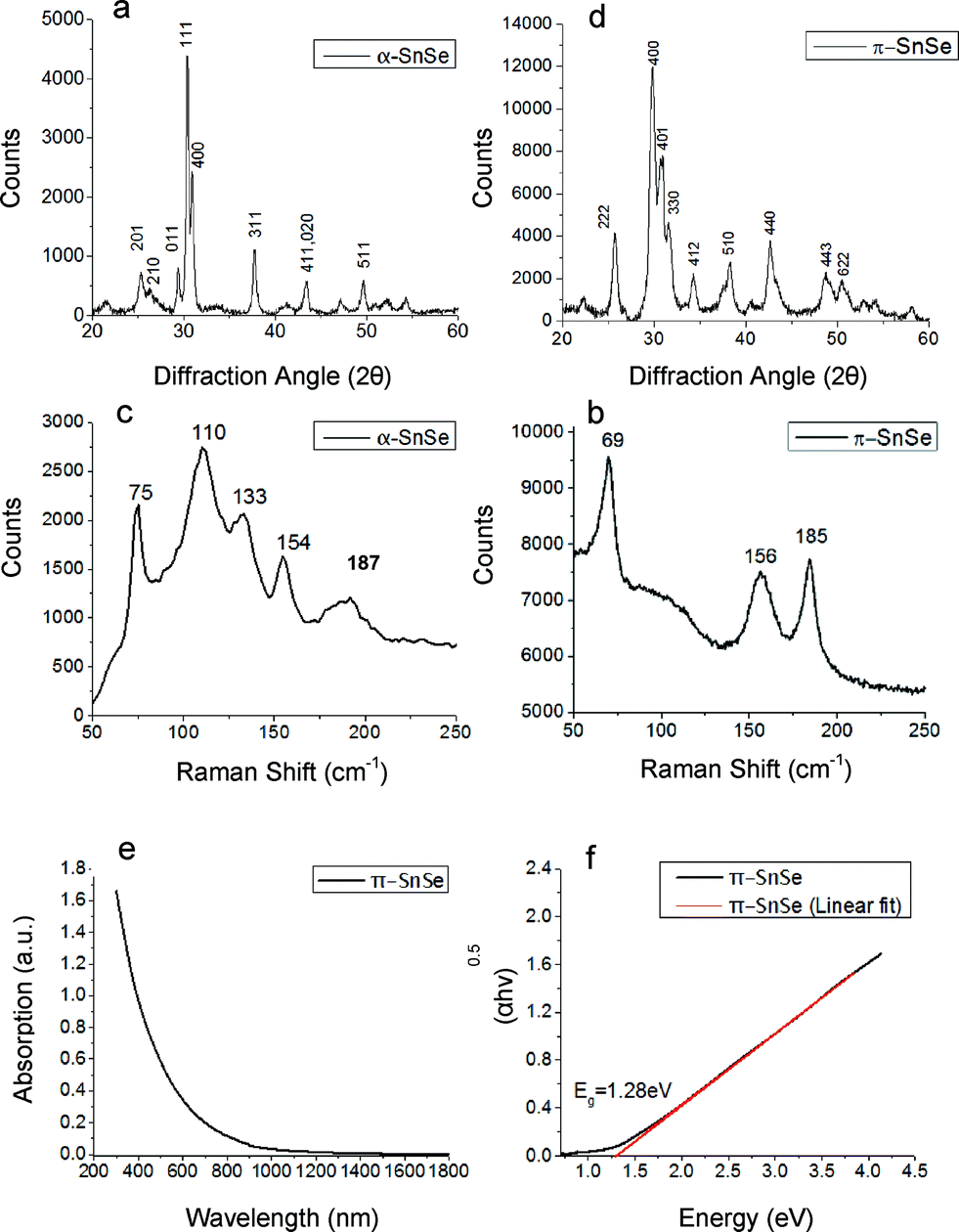 | ||
| Fig. 2 Powder XRD patterns corresponding to (a) α-SnSe with irregular morphologies (b) π-SnSe with cube-like morphology. Raman spectra corresponding to (c) α-SnSe nanoparticles (d) π-SnSe nanoparticles. The new π-SnSe phase shows less features than conventional α-SnSe in the investigated region. (e) UV-vis-NIR spectra of π-SnSe cube-like nanoparticles. (f) Tauc plot of the corresponding absorption spectra, indicating a band gap of 1.28 eV for the π-SnSe phase. | ||
High resolution TEM (HRTEM) micrographs of two cube-like nanoparticles at different zone axes (ZA) are presented in Fig. 1e and f; the inset in Fig. 1e and f shows the corresponding fast Fourier transform (FFT) pattern. The FFT patterns are in agreement with the dhkl-spacing extracted from SAED patterns. From the analysis of the net symmetry presented in these FFT patterns, it was plausible to assume that the π-SnSe crystallized in a cubic symmetry with a0 = 12.1 ± 0.04 Å.
In a recent report from our group, we have used the precession electron diffraction tomography (PEDT) technique for obtaining a full structure solution for the π-SnS phase (P213, a0 = 11.7 Å), a previously unknown simple cubic binary phase of tin mono-sulfide.28 Significant progress in the synthesis allowed us to obtain pure powder samples of the π-SnS nanocrystalline phase. Subsequently, refinement of the structural model was carried out using Rietveld refinement on powder X-ray diffraction data, establishing a refined lattice parameter of a0 = 11.595 Å.31 Moreover, the model also confirms with solution deposited thin film samples of π-SnS.32,33 Assuming that the tin mono-selenide phase belong to the π-SnS structural type, both electron diffraction data and XRD could be indexed appropriately. By replacing S with Se atom type, a theoretical π-SnSe structural model was proposed. Then, applying the Rietveld method utilized in FULLPROF software,34 the theoretical π-SnSe model was fitted to the experimental powder XRD data. Negligible difference between calculated and observed X-ray diffraction profiles along with low values of reliability factors (Rp = 1.65 Rwp = 2.11), obtained at the final Rietveld π-SnSe structure refinement, confirmed the applicability of the structural solution of the π-SnS for the π-SnSe phase. The results are presented in detail in Fig. S1 in ESI.† The refined lattice parameter of π-SnSe was determined to be a0 = 11.9702(9) Å. XRD and electron diffraction in TEM are different techniques for lattice parameter determination. Thus, deviation in the results can be expected (a0 = 11.9702(9) Å in XRD vs. a0 = 12.1 in TEM Å, a 1.07% difference). Yet, excellent match has been achieved by different techniques. We note that XRD is the most accurate technique for determination of the lattice parameter of powder samples.
Comparison of the relative atomic positions of π-SnSe with those of π-SnS shows that they do not differ from one other by more than a few percent, with the lattice parameters scaling similarly to within less than 3.2%. These variations are well expected since the selenide anion is larger than the sulfide anion. As discussed above, SnS and SnSe are part of the IV–VI family of semiconductor materials, and share similarities with GeS and GeSe, namely belong to the orthorhombic GeS prototype. The discovery of π-SnS and π-SnSe could point toward a new simple cubic prototype in this system, yet no similar simple cubic phase for GeS or GeS has been reported to date. We also note that the new cubic phase, π-SnSe belongs to a class of non-centrosymmetric crystals. This, in turn, gives rise to piezoelectricity, optical activity and enantiomorphism – an important and potentially useful material properties dictated by crystal symmetry. While this warrants further consideration, both experimental and theoretical, we note that these features are not expected for the orthorhombic crystal structure.
Since our synthetic protocol allows synthesizing each SnSe phase separately, we have explored the properties of π-SnSe using macroscopic characterization tools. Fig. 2 shows the Raman spectra of both π-SnSe and α-SnSe. The α-SnSe spectrum shows peaks at 75 cm−1, 110 cm−1, 133 cm−1, 154 cm−1, 187 cm−1. The 110 cm−1 peak corresponds to the B3g mode, while the peaks at 75 cm−1, 133 cm−1, 154 cm−1 belong to Ag phonon modes, all attributed to α-SnSe.7,35–37 The peak at 185 cm−1 belongs to SnSe2.38 It was found that both α-SnSe and π-SnSe particles are sensitive to the excitation laser beam, which causes them to transform from SnSe to SnSe2 over time. The Raman spectrum of π-SnSe shows only 3 sharp peaks in the same region, 69 cm−1, 156 cm−1 and 185 cm−1. Another weak and broad peak is centered around 120 cm−1. The peak at 185 cm−1 is again attributed to SnSe2 while the other peaks are assigned to π-SnSe. Comparing the two spectra shows that the π-SnSe 110 cm−1 and 133 cm−1 peaks are substantially weakened. Such changes are expected due to the different structure, and the subsequent lattice symmetry of the two phases. The 185 cm−1 peak is sharper in the spectrum of π-SnSe. We speculate that the larger surface area in π-SnSe nanoparticles makes them more sensitive to the laser beam, causing them to readily transform from π-SnSe to SnSe2. Surprisingly, comparison of the Raman spectra of α-SnS and π-SnS showed quite a different picture. While π-SnS showed several additional peaks compared to α-SnS,31Fig. 2d shows restraining of phonons in the Raman spectrum of the π-SnSe phase as compared vs. α-SnSe. This phenomenon of phonon restraining could be related to the change in the tin and selenide coordination. It is well known that upon heating the orthorhombic phase of SnSe, a second order phase transition occurs from Pnma (space group no. 62) to Cmcm (space group no. 63). This transition could be described as a change in the coordination polyhedron of the tin and selenide ions, with higher symmetry in the high temperature orthorhombic phase. The higher site symmetry results in phonon anharmonicity and substantial phonon scattering.39 Examination of the discussed cubic crystal structure with respect to the low temperature orthorhombic structure suggests that a similar transition may be occurring. In this case, both tin and selenide ions are located in “4a” Wyckoff sites (space group no. 198) with each ion surrounded by three ions of the opposite valence in 3-fold symmetry. Thus, the coordination polyhedron for both tin and selenium ions could be represented as a pyramidal sub-structure in the π-SnSe crystal. We speculate that this unique arrangement could adversely affect phonon propagation in the material, similarly to the previously proposed mechanism.39 This mechanism of phonon anharmonicity has been reported to be responsible for the ultra-low thermal conductivity of α-SnSe at high temperatures.29,39 Thus, we conclude that while substantial phonon scattering is observed in orthorhombic α-SnSe only at high temperature, the new cubic π-SnSe is expected to exhibit significant phonon scattering also at room temperature. Further studies are underway in order to explore these phenomena and to assign the specific phonon modes to the peaks in the Raman spectrum of π-SnSe.
The optical properties of π-SnSe were explored using UV-vis-NIR absorption spectroscopy. The π-SnSe absorption curves and the corresponding Tauc plot for π-SnSe cube-like nanoparticles are shown in Fig. 2. We note that the literature reported band of α-SnSe is 0.9 eV (indirect) and 1.3 eV (direct).6,7,9,10,30,37 The absorption curve in Fig. 2e shows an onset of absorption around ∼950 nm. The band gap of an indirect bandgap semiconductor is related to the absorption coefficient α according to the formula αhv0.5 = A(hv − Eg) where h is Planck's constant, v is the light frequency, Eg is the material band gap and A is a constant. The band gap can be obtained from the x-intercept upon plotting (αhv)0.5vs. hv, as shown in Fig. 2f. The plot yielded a straight line, indicating that π-SnSe is an indirect band gap material, with the extrapolated fitted line intercepting the hv axis at 1.28 eV. Thus, the bandgap of π-SnSe is significantly larger than that of α-SnSe, with a difference of about 0.4 eV. This implies that while α-SnSe is active in the short wave infrared range, the new π-SnSe phase is expected to be active closer to the visible range, with technological implications for solar cells, thermoelectric applications and photocatalysis.1,2,40,41 A similar increase was reported also for π-SnS with respect to α-SnS, where the band gap is increased from 1.07 eV in conventional α-SnS to 1.53–1.72 eV in π-SnS.31–33
The stable phase of tin selenide in the solid state is the 8 atom orthorhombic α-SnSe with the space group Pnma. We performed DFT-PBE calculations of this orthorhombic 8 atom phase and of the rocksalt phase for reference purposes. For the orthorhombic phase, the calculated lattice parameters were found to be a0 = 11.8346 Å, b0 = 4.2254 Å, c0 = 4.54 Å, compared to the experimental values of JCPDS no. 48-1224, a0 = 11.498 Å, b0 = 4.153 Å, c0 = 4.440 Å. Representing deviations of up to several percent as might be expected. The electronic band structure in the relaxed position was obtained and is presented in Fig. 3a.
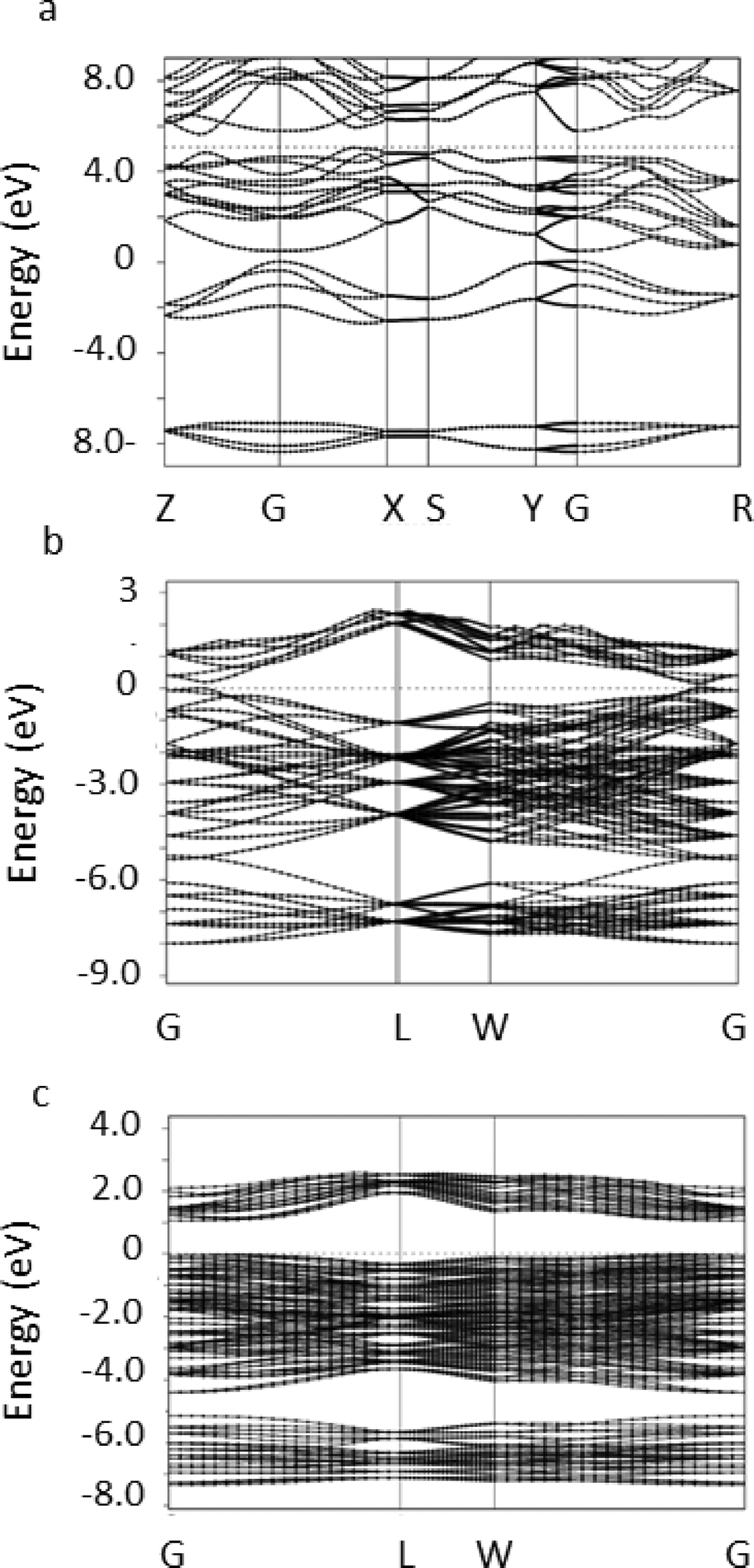 | ||
| Fig. 3 (a) Band structure of the orthorhombic α-SnSe phase, with 8 atoms in the Herzenbergite structure. (b) Calculated band structures of the ideal rock salt lattice with 64 atoms per unit cell (c) and the π-SnSe phase with 64 atoms per unit cell. | ||
It can be seen that an indirect band-gap of ca. 0.597 eV is found between the points Z and X and a direct band-gap of ca. 0.867 eV is found between the high symmetry points Z and G, in excellent, but probably fortuitous, agreement with experimental measurements of 0.9 eV.7,9,10,30,37
To study the 64 atom π-SnSe phase we constructed a simple cubic unit cell consisting of the 64 atoms initially located in their experimentally determined positions. A structural relaxation allowing the atomic positions, the volume and the lattice constants of the unit cell to vary was then performed. In this cell, convergence of the calculation was achieved with a 8 × 8 × 8 k-point mesh. A total energy of −829.8196 Ry per 64 atom unit cell, a total (isotropic) stress of −0.32 kbar and a relaxed lattice parameter of 12.284 Å which is slightly larger than the experimental value of 11.9702(9) Å reported above indicating that the phase is mechanically stable. The stability of the π-SnSe phase can be examined by comparing the total energy per atom pair of −25.9319 Ry to −25.9342 Ry in the ideal rocksalt phase and −25.9339 Ry in the orthorhombic phase. We see that the total energy results predict the π-SnSe phase to be less stable than the orthorhombic phase by approximately 2 × 10−3 Ry per atom which is computationally significant and at the limit of physical significance. The orthorhombic α-SnSe phase is slightly less stable in our calculation than the energetically equivalent rocksalt phase, but this difference is physically insignificant and of the same order of magnitude as found in earlier studies.42
To examine the origin of the stability of the π-SnSe phase relative to the ideal rocksalt structure we examined the band structure of both systems in a unit cell containing 64 atoms, i.e. with a similar size of the Brillouin zone. The results are shown in Fig. 3c. It is immediately seen that the band structure in the π-SnSe phase is distorted relative to the highly symmetric structure in the ideal rocksalt phase, as can be seen, e.g., at the L point. More significantly, the energy gap between the valence and conduction band is found to increase from ca. 0.2 eV in the ideal structure to approximately 1.18 eV in the π-SnSe phase in very good agreement with the measurements reported above (1.28 eV). We note that DFT-PBE is well-known to underestimate band gaps.43 Due to the large size of the unit cell, and the consequently small size of the Brillouin zone, it was impossible to determine from our calculations whether the band gap is indirect.
Conclusions
We have reported on a new binary phase of tin mono-selenide, π-SnSe. A proposed theoretical π-SnSe model, based on the recently discovered π-SnS structure was constructed by replacing S ion with Se ions. Using this modified model, we successfully indexed both XRD and electron diffraction patterns obtained for the new π-SnSe material. Rietveld refinement of the modified π-SnSe model was carried on XRD data. The high compatibility between the modified π-SnSe model and observed XRD profiles, along with low reliability factors, provided strong proof for the correctness of the π-SnSe model, and points towards a new prototype which may exist in parallel to the GeS prototype. Since the π-SnSe phase was obtained as a single phase of pure cube-like nanoparticle powder, it was possible to carry out Raman spectroscopy and UV-vis-NIR absorption spectroscopy. Raman spectroscopy showed a distinct fingerprint for π-SnSe, with restrained phonons (smaller number of peaks) in comparison to α-SnSe. This phenomenon could be potentially useful for thermoelectric applications where low thermal conductivity is desired. UV-vis-NIR spectroscopy indicated an indirect band gap of ca. 1.28 eV, which is larger by about 0.4 eV with respect to that of α-SnSe (0.9 eV, indirect). Due to the different crystallographic structure, this new cubic phase of SnSe is predicted to exhibit interesting and potentially useful piezoelectric properties, unlike the conventional orthorhombic phase. DFT total energy calculations indicated that the π-SnSe is mechanically stable and is energetically close to the stable α-SnSe phase. The band gap is confirmed to increase from ca. 0.9 to 1.2 eV in very good, but possibly fortuitous, agreement with the experimental measurements.Acknowledgements
We thank Dr. Dimitry Mogilyanski for expert assistance with XRD, Dr. Iddo Pinkas for expert assistance with Raman spectroscopy and Eyal Toutian for assistance with nanoparticle synthesis. This work was supported by the Israel Science Foundation under Grant # 340/2010, and by the Focal Technological Area (FTA) Program of the Israel National Nanotechnology Initiative (INNI).References
- L. M. Peter, Philos. Trans. R. Soc., A, 2011, 369, 1840–1856 CrossRef CAS PubMed
.
- S. Abermann, Sol. Energy, 2013, 94, 37–70 CrossRef CAS
- C. Gammer, C. Mangler, C. Rentenberger and H. P. Karnthaler, Scr. Mater., 2010, 63, 312–315 CrossRef CAS
- B. K. Patra, S. Sarkar, A. K. Guria and N. Pradhan, J. Phys. Chem. Lett., 2013, 4, 3929–3934 CrossRef CAS
- J. Ning, K. Men, G. Xiao, L. Wang, Q. Dai, B. Zou, B. Liu and G. Zou, Nanoscale, 2010, 2, 1699–1703 RSC
- J. J. Ning, G. J. Xiao, T. Jiang, L. Wang, Q. Q. Dai, B. Zou, B. B. Liu, Y. J. Wei, G. Chen and G. T. Zou, CrystEngComm, 2011, 13, 4161–4166 RSC
- S. L. Zhao, H. A. Wang, Y. Zhou, L. Liao, Y. Jiang, X. Yang, G. C. Chen, M. Lin, Y. Wang, H. L. Peng and Z. F. Liu, Nano Res., 2015, 8, 288–295 CrossRef CAS
- S. G. Hickey, C. Waurisch, B. Rellinghaus and A. Eychmuller, J. Am. Chem. Soc., 2008, 130, 14978–14980 CrossRef CAS PubMed
- L. Li, Z. Chen, Y. Hu, X. Wang, T. Zhang, W. Chen and Q. Wang, J. Am. Chem. Soc., 2013, 135, 1213–1216 CrossRef CAS PubMed
- D. D. Vaughn 2nd, S. I. In and R. E. Schaak, ACS Nano, 2011, 5, 8852–8860 CrossRef PubMed
- P. Ramasamy, P. Manivasakan and J. Kim, CrystEngComm, 2015, 17, 807–813 RSC
- X. Liu, Y. Li, B. Zhou, X. Wang, A. N. Cartwright and M. T. Swihart, Chem. Mater., 2014, 26, 3515–3521 CrossRef CAS
- Z. Deng, D. Cao, J. He, S. Lin, S. M. Lindsay and Y. Liu, ACS Nano, 2012, 6, 6197–6207 CrossRef CAS PubMed
- A. J. Biacchi, D. D. Vaughn 2nd and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 11634–11644 CrossRef CAS PubMed
- A. de Kergommeaux, M. Lopez-Haro, S. Pouget, J. M. Zuo, C. Lebrun, F. Chandezon, D. Aldakov and P. Reiss, J. Am. Chem. Soc., 2015, 137, 9943–9952 CrossRef CAS PubMed
- K. Ramasamy, V. L. Kuznetsov, K. Gopal, M. A. Malik, J. Raftery, P. P. Edwards and P. O'Brien, Chem. Mater., 2013, 25, 266–276 CrossRef CAS
- G. H. Yue, Y. D. Lin, X. Wen, L. S. Wang and D. L. Peng, J. Mater. Chem., 2012, 22, 16437–16441 RSC
- D. D. Vaughn 2nd, O. D. Hentz, S. Chen, D. Wang and R. E. Schaak, Chem. Commun., 2012, 48, 5608–5610 RSC
- L. L. Cheng, M. H. Liu, M. X. Wang, S. C. Wang, G. D. Wang, Q. Y. Zhou and Z. Q. Chen, J. Alloys Compd., 2012, 545, 122–129 CrossRef CAS
- W. Cai, J. Hu, Y. S. Zhao, H. L. Yang, J. Wang and W. D. Xiang, Adv. Powder Technol., 2012, 23, 850–854 CrossRef CAS
- Y. Zhang, J. Lu, S. Shen, H. Xu and Q. Wang, Chem. Commun., 2011, 47, 5226–5228 RSC
- E. C. Greyson, J. E. Barton and T. W. Odom, Small, 2006, 2, 368–371 CrossRef CAS PubMed
- D. Avellaneda, M. T. S. Nair and P. K. Nair, J. Electrochem. Soc., 2008, 155, D517–D525 CrossRef CAS
- A. Akkari, C. Guasch and N. Kamoun-Turki, J. Alloys Compd., 2010, 490, 180–183 CrossRef CAS
- Z. Deng, D. Han and Y. Liu, Nanoscale, 2011, 3, 4346–4351 RSC
- I. Y. Ahmet, M. S. Hill, A. L. Johnson and L. M. Peter, Chem. Mater., 2015, 27, 7680–7688 CrossRef CAS
- L. A. Burton and A. Walsh, J. Phys. Chem. C, 2012, 116, 24262–24267 CAS
- A. Rabkin, S. Samuha, R. E. Abutbul, V. Ezersky, L. Meshi and Y. Golan, Nano Lett., 2015, 15, 2174–2179 CrossRef CAS PubMed
- L. D. Zhao, S. H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef CAS PubMed
- W. J. Baumgardner, J. J. Choi, Y. F. Lim and T. Hanrath, J. Am. Chem. Soc., 2010, 132, 9519–9521 CrossRef CAS PubMed
- R. E. Abutbul, E. Segev, L. Zeiri, V. Ezersky, G. Makov and Y. Golan, RSC Adv., 2016, 6, 5848–5855 RSC
- P. K. Nair, A. R. Garcia-Angelmo and M. T. S. Nair, Phys. Status Solidi A, 2016, 213, 170–177 CrossRef CAS
- A. R. Garcia-Angelmo, R. Romano-Trujillo, J. Campos-Álvarez, O. Gomez-Daza, M. T. S. Nair and P. K. Nair, Phys. Status Solidi A, 2015, 212, 2332–2340 CrossRef CAS
-
J. Rodrigues-Carvajal, Program FULLPROF-98, Version 5.06, 2015 Search PubMed
- H. R. Chandrasekhar, R. G. Humphreys, U. Zwick and M. Cardona, Phys. Rev. B: Solid State, 1977, 15, 2177–2183 CrossRef CAS
- M. Parenteau and C. Carlone, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 4, 5227–5234 CrossRef
- J. Zhang, H. Y. Zhu, X. X. Wu, H. Cui, D. M. Li, J. R. Jiang, C. X. Gao, Q. S. Wang and Q. L. Cui, Nanoscale, 2015, 7, 10807–10816 RSC
- D. G. Mead and J. C. Irwin, Solid State Commun., 1976, 20, 885–887 CrossRef CAS
- C. W. Li, J. Hong, A. F. May, D. Bansal, S. Chi, T. Hong, G. Ehlers and O. Delaire, Nat. Phys., 2015, 11, 1063–1069 CrossRef CAS
- Z. Zhang, W. Wang, L. Wang and S. Sun, ACS Appl. Mater. Interfaces, 2012, 4, 593–597 CAS
- S. B. Rawal, S. Bera, D. Lee, D. J. Jang and W. I. Lee, Catal. Sci. Technol., 2013, 3, 1822–1830 CAS
- A. Walsh and G. W. Watson, J. Phys. Chem. B, 2005, 109, 18868–18875 CrossRef CAS PubMed
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2009, 130, 074103 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Methods and characterization techniques, Rietveld refinement data, crystallographic information file (CIF). See DOI: 10.1039/c5ce02437d |
| This journal is © The Royal Society of Chemistry 2016 |