DOI:
10.1039/C5CE02282G
(Paper)
CrystEngComm, 2016,
18, 948-961
Two modes of peri-interaction between an aldehyde group and a carboxylate anion in naphthalaldehydate salts†
Received
23rd November 2015
, Accepted 31st December 2015
First published on 4th January 2016
Abstract
Crystal structures of the salts of 1,8-naphthalaldehydic acid (8-formyl-1-naphthoic acid) show one of two types of interaction between the functional groups. In the more commonly observed case, a carboxylate oxygen lies close to the aldehyde carbonyl carbon atom (O⋯C: 2.445–2.630 Å) and makes an n–pi* interaction. However, in two other cases the carboxylate group has rotated so that the aldehyde now directs its hydrogen atom at the face of the carboxylate group and forms a surprisingly short contact with the carbon atom (H⋯C: 2.29 and 2.42 Å). This interaction is likely to be electrostatic in nature.
Introduction
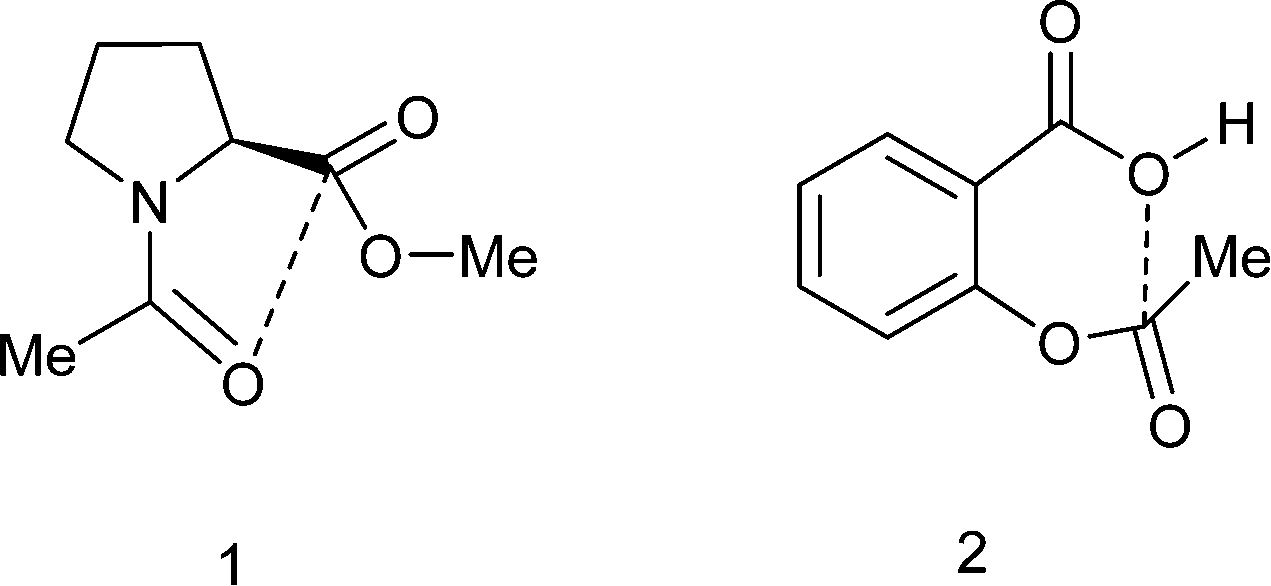
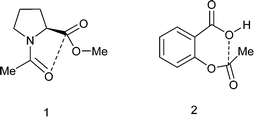
Carbonyl/carbonyl interactions of type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO, in which a sp2 oxygen lone pair impinges on a carbonyl carbon, can be regarded as n–π* interactions and have been proposed to stabilise the conformations of proteins,1 polymers2 and small molecules such as substituted N-acetyl-proline methyl esters 1 (Scheme 1).3,4 The interaction leads to a small pyramidalisation of the carbonyl carbon involved. Interestingly, it has been proposed that such interactions can provide a link in the relay of chirality in a protein,5 and there is evidence that the formation of such an interaction modifies the properties of the groups involved.4 Furthermore, in the crystal structure of aspirin 2 a similar interaction is observed but between the hydroxyl oxygen of the carboxylic acid and the ester carbonyl group (Scheme 1). Calculations on aspirin and its anion support the existence of such stabilising interactions, which are larger for the anion, and which may be responsible for the high acidity of aspirin, as well as its efficacy as an acyl transfer agent in biology.6 The O⋯C distances from neutral oxygen observed in these interactions are typically >2.8 Å and calculations suggest stabilisations varying from 0.9 kcal mol−1 in aspirin to about 0.27 kcal mol−1 between two amide groups.
O⋯CO, in which a sp2 oxygen lone pair impinges on a carbonyl carbon, can be regarded as n–π* interactions and have been proposed to stabilise the conformations of proteins,1 polymers2 and small molecules such as substituted N-acetyl-proline methyl esters 1 (Scheme 1).3,4 The interaction leads to a small pyramidalisation of the carbonyl carbon involved. Interestingly, it has been proposed that such interactions can provide a link in the relay of chirality in a protein,5 and there is evidence that the formation of such an interaction modifies the properties of the groups involved.4 Furthermore, in the crystal structure of aspirin 2 a similar interaction is observed but between the hydroxyl oxygen of the carboxylic acid and the ester carbonyl group (Scheme 1). Calculations on aspirin and its anion support the existence of such stabilising interactions, which are larger for the anion, and which may be responsible for the high acidity of aspirin, as well as its efficacy as an acyl transfer agent in biology.6 The O⋯C distances from neutral oxygen observed in these interactions are typically >2.8 Å and calculations suggest stabilisations varying from 0.9 kcal mol−1 in aspirin to about 0.27 kcal mol−1 between two amide groups.
 |
| | Scheme 1 n–π* interactions between oxygen and carbonyl groups. | |
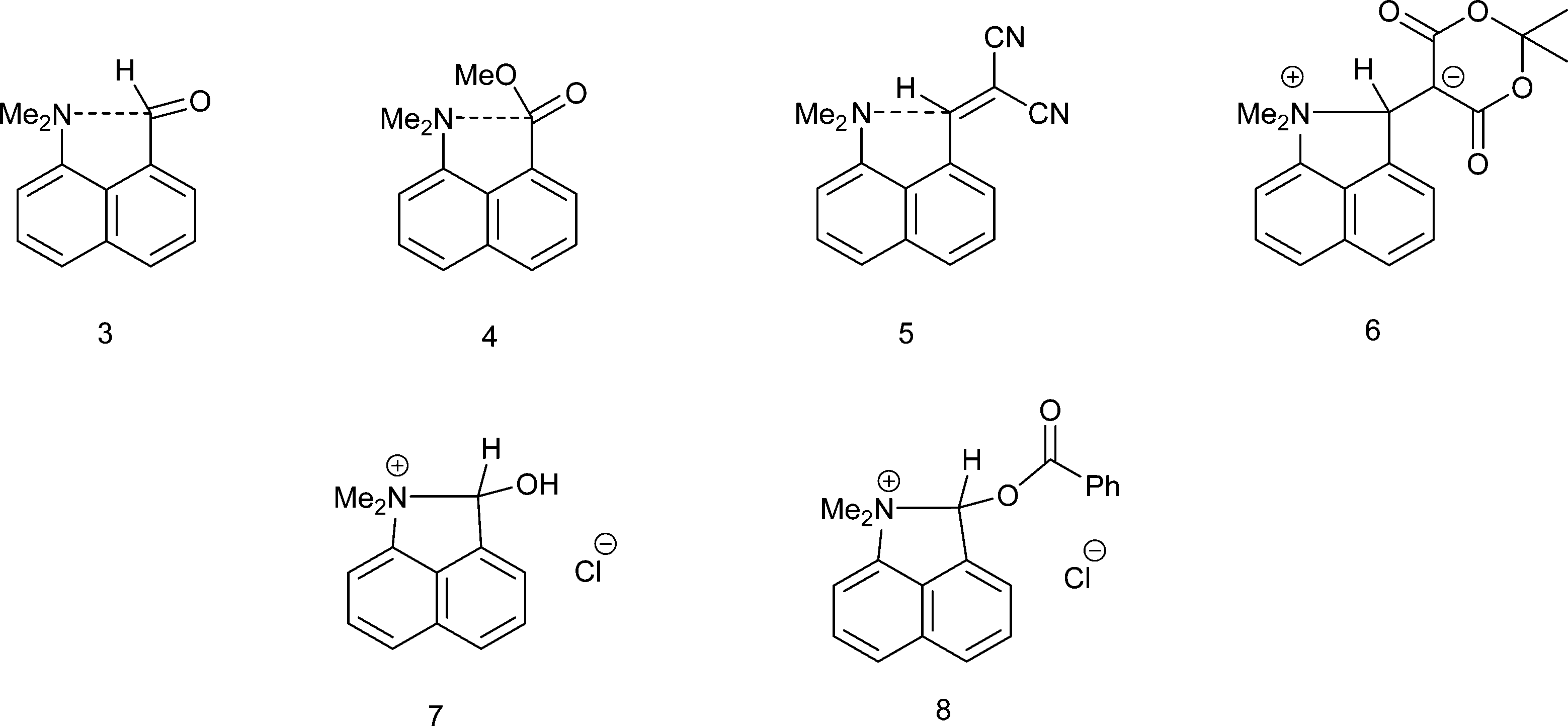
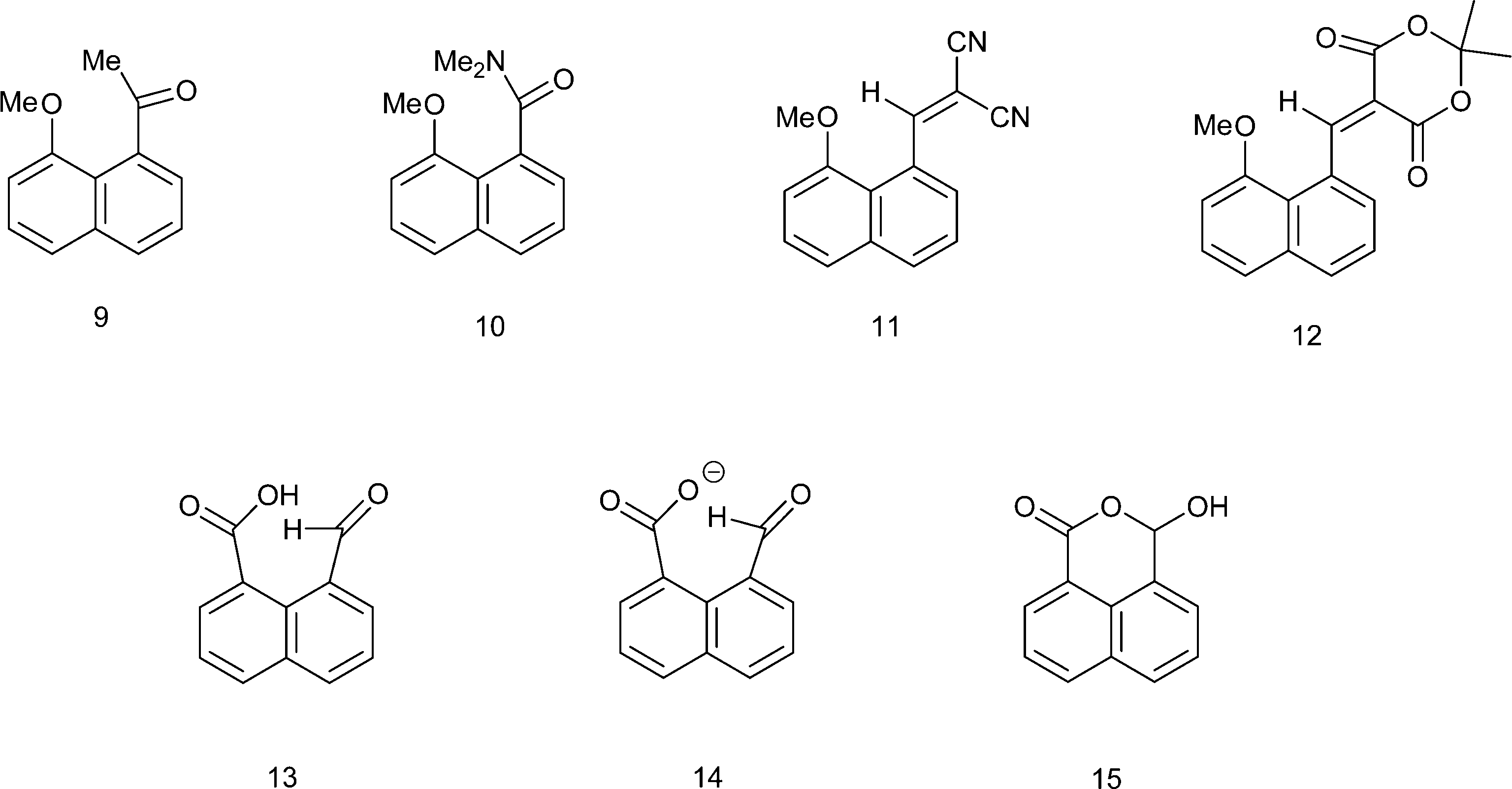
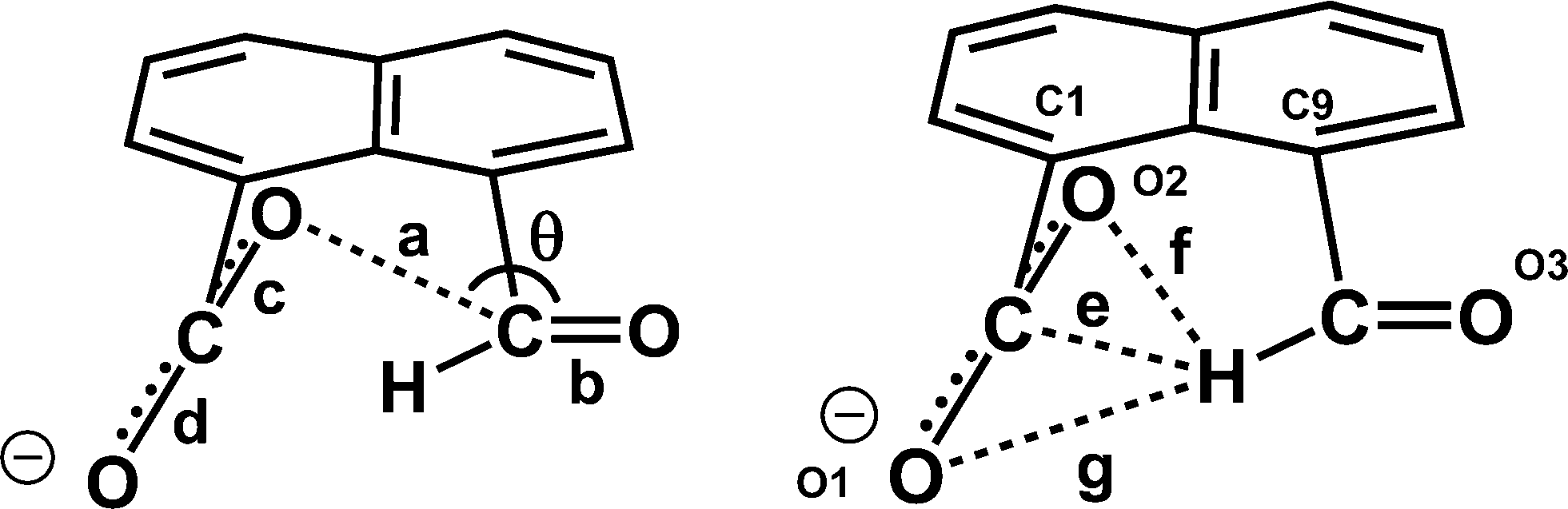
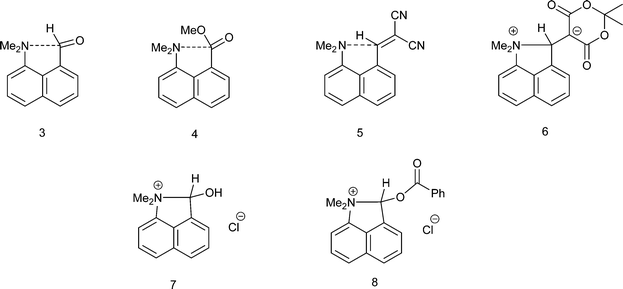
We have been investigating the interactions and reactions of the more nucleophilic dimethylamino group with various electrophilic organic groups placed in the peri positions of the naphthalene ring system. This work, initiated by Dunitz et al. on studies with carbonyl containing electrophiles as in 3 and 4,7 has been extended to electron deficient alkenes e.g.5 some of which react with the peri-dimethylamino group to produce zwitterions with long C–N bonds between the groups as in compound 6 (Scheme 2).8–10 This approach has been extended to the biphenyl and triptycene scaffolds.11,12 Together with several compounds containing short transannular N⋯C interactions13 a set of structures with N⋯C separations over the range 2.764 to 1.612 Å has been built up which represent different stages in the formation of the N–C bond. The shorter N⋯C distances in this range correspond to long covalent N–C bonds. The longer N⋯C separations observed for interactions with esters and amides can be considered as n–π* interactions corresponding to early stages in bond formation and are associated with small pyramidalisations of the carbonyl carbon towards the nucleophile. Such an interaction may be the initiation for further chemical reactions. Thus, the aldehyde 3 reacts readily on oxygen with a proton or an acyl group at 0 °C with concomitant formation of a N–C bond between peri-substituents to give 710 and 8,14 and similar protonations have been observed with some analogous ketones.15 In contrast, when the dimethylamino group is replaced by the much less nucleophilic methoxy group there is very little change in the MeO⋯C separations in a similar range of peri-naphthalenes 9–12 (2.550–2.611 Å), though there are small pyramidalisations of the adjacent ketone or amide groups towards the methoxy group (0.039–0.044 Å) (Scheme 3).16 Outside the peri-naphthalene series we have also observed similar contacts between an ether oxygen and a ketone,12 and between an ester carbonyl oxygen and a nitrile, leading to small distortions of the electrophilic functional group.17
 |
| | Scheme 2 Interactions and bond formations between substituents in peri-naphthalenes. | |
Given the weak nature of interactions involving neutral oxygen, we decided to investigate the electrophile/nucleophile interactions involving the more nucleophilic negatively charged oxygen, and here we report the structures of a series of salts of 1,8-naphthalaldehydic acid (8-formyl-1-naphthoic acid) 13 in whose anion 14 a negatively charged carboxylate group could make a close approach to an aldehyde group. Such interactions would be the anionic equivalent of the n–π* interactions between carbonyl groups described above. The parent acid adopts a cyclic structure 15 in the solid and solution state, so that in this case the groups have interacted fully and formed a bond (Scheme 3).
 |
| | Scheme 3
Peri-naphthalenes containing methoxy or carboxylate groups as the nucleophilic group. | |
Discussion
1,8-Naphthalaldehydic acid – cyclic form 15
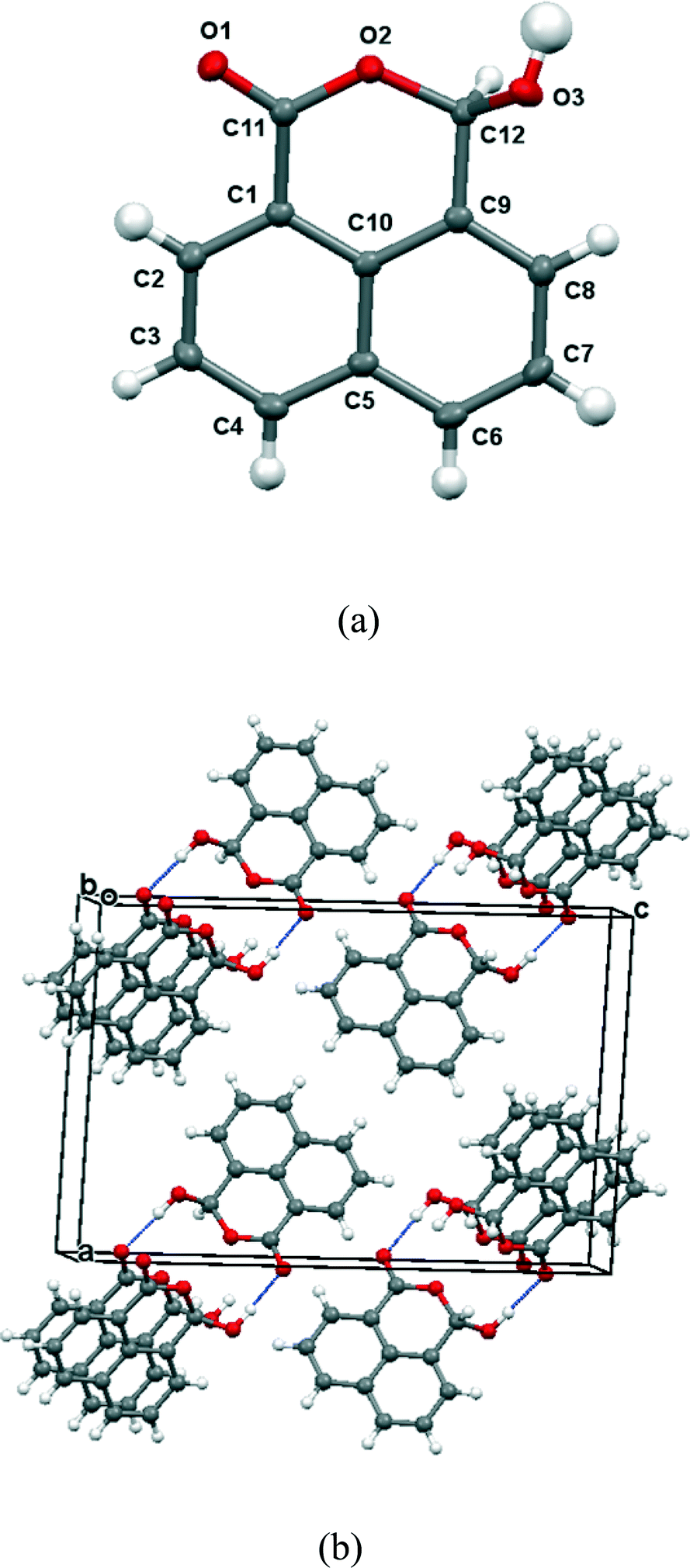
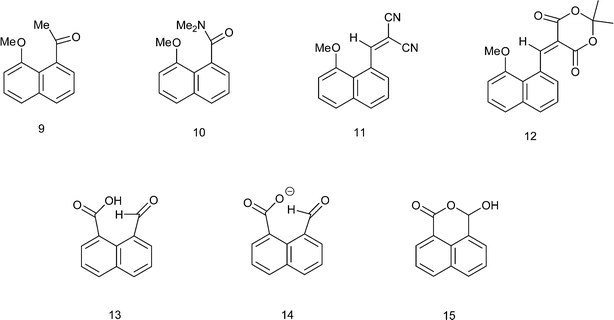
The 13C NMR spectrum of naphthalaldehydic acid dissolved in (CD3)2CO was consistent with the cyclic structure 15 where the carboxylic acid group has added to the aldehyde, showing shifts of δH: 6.94 and δc: 97.1 for the methine group attached to two oxygen atoms. Similar results were obtained from solution in CD3OD. To investigate the solid state structure of 1,8-naphthalaldehydic acid, crystals were grown from acetonitrile and the structure determined by X-ray diffraction at 100 K. The crystal structure, which is monoclinic and in space group P21/c, confirmed that the cyclic structure is also present in the solid state (Fig. 1). Thus, a bond has been formed between the carboxylic acid and aldehyde groups (1.477(2) Å). This bond experiences a significant anomeric effect from the hydroxyl group causing it to lengthen from the typical value for a six-ring lactone (ca. 1.34 Å)18 and shortening the C–OH bond from a typical value of ca. 1.43 Å18 to 1.387(2) Å. The molecules are stacked along the b axis with O–H⋯OC hydrogen bonds between molecules in adjacent stacks related by a two-fold axis (O3–H3A: 0.94(2), H3A⋯O1′: 1.84(2), O3⋯O1′ 2.785(4) Å, O3–H3A⋯O1′: 176.5(17)°). In contrast, the crystal structures of the salts of naphthalaldehydic acid show that the anion does not adopt a cyclic structure which would have to contain an alkoxy anion, but consists of a naphthalene system with the more stable carboxylate anion and aldehyde group as peri substituents.
 |
| | Fig. 1 (a): Molecular structure for 1,8-naphthalaldehydic acid in its cyclic form 15. Selected molecular geometry: O(1)–C(11): 1.2226(19), O(2)–C(11) 1.341(2), O(2)–C(12): 1.477(2), O(3)–C(12): 1.387(2) Å, O(2)–C(12)–O(3): 106.67(12)°; (b) crystal packing arrangement for 15 showing hydrogen bonding linking molecules along the b axis. | |
Group 1A salts of the naphthalaldehydate anion 14
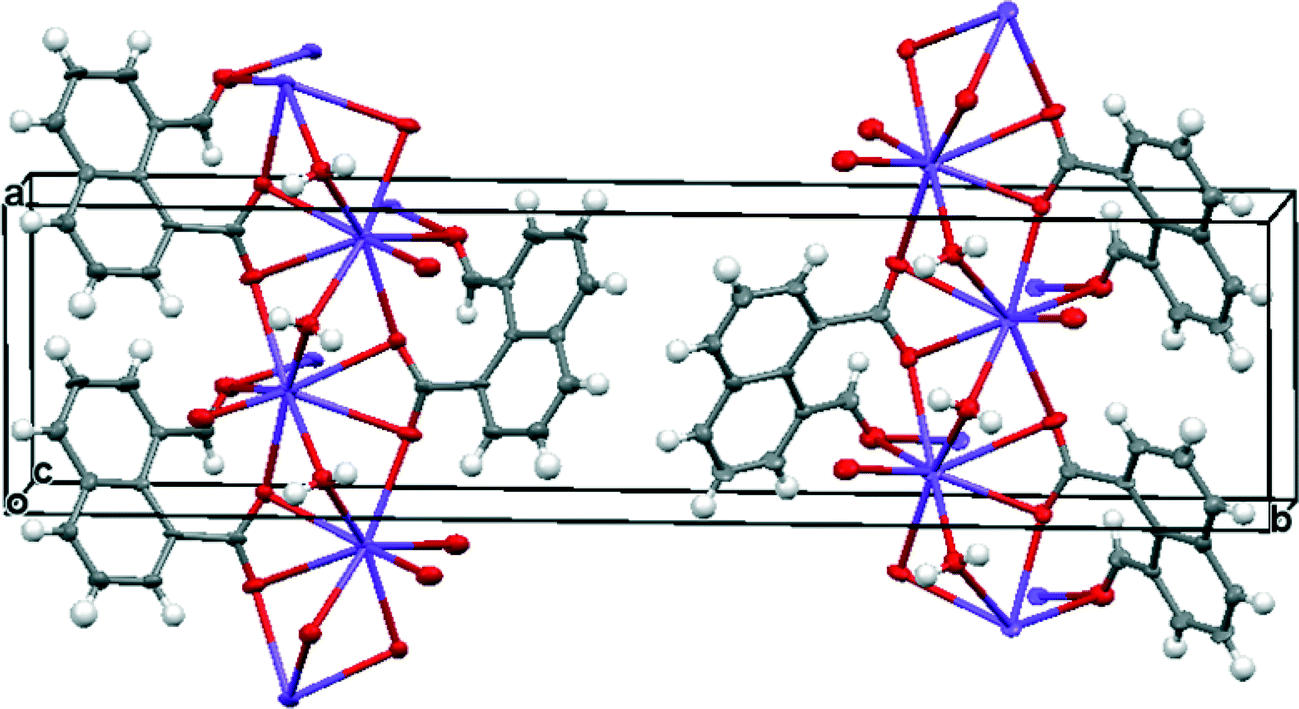
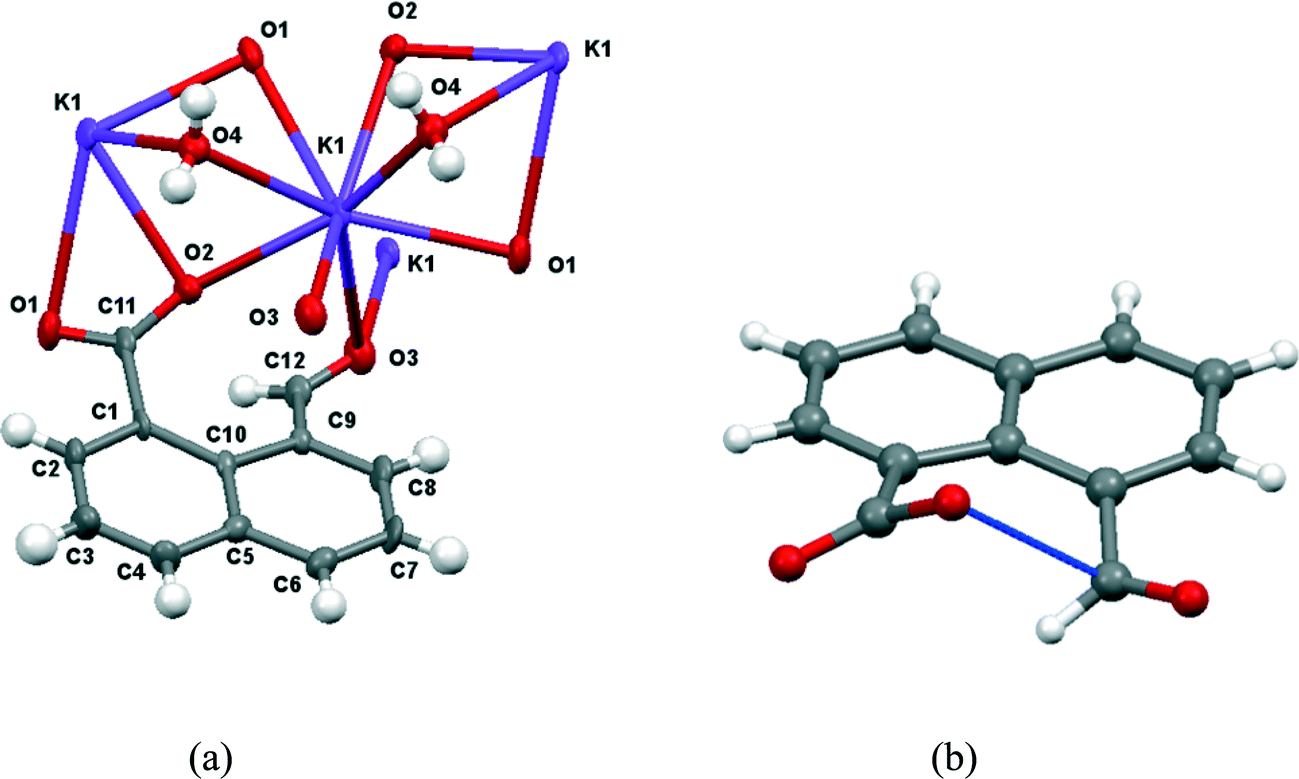
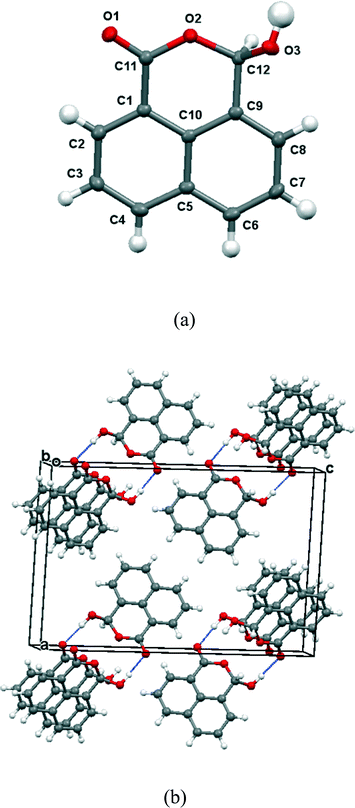
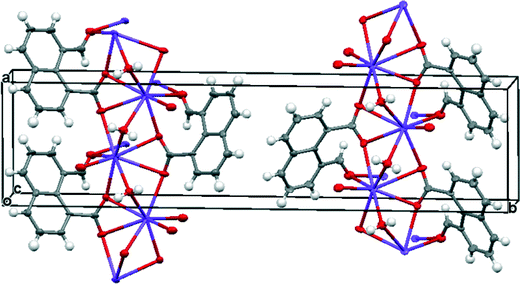
Group 1A salts of 1,8-naphthalaldehydic acid were prepared by reaction with the corresponding metal hydroxides to give the lithium, potassium and cesium salts and crystals were grown from various alcohols. The potassium and cesium salts crystallise as monohydrates and are isostructural with orthorhombic crystal structures in space group Pna21. The metal ions are coordinated by eight oxygen atoms: two sets of carboxylate groups, two aldehyde groups and two water molecules. Each carboxylate oxygen atom and the aldehyde oxygen atom are all coordinated to two metal ions and the water molecule bridges two metal ions. The functional groups are oriented so that their planes lie at 32° to each other and one carboxylate oxygen O(2) approaches the aldehyde carbon atom, and the aldehyde hydrogen atom lies nearly over the carboxylate group. The crystal structure of the potassium salt is shown in Fig. 2, and the interactions involving the anion are shown in Fig. 3. For the potassium salt, the carboxylate group is unsymmetrical with carbon–oxygen bonds of 1.241(10) (to O1) and 1.280(9) Å (to O2) suggesting that more negative charge is concentrated on O2. Apart from the interaction with the aldehyde, this oxygen also makes two short coordinations to potassium ions (2.810(6) and 2.863(6) Å) while for O(1) one of its two coordinations to potassium ions is longer (2.872(6) and 3.140(7) Å) which will also influence the C–O bond lengths. The carboxylate and aldehyde groups are rotated out of the naphthalene plane in the same sense by similar amounts (33.3 and 29.2° respectively), splayed apart in-plane and displaced to opposite sides of the naphthalene's best plane so that the distance between the more negatively charged carboxylate oxygen atom O(2) and the aldehyde carbon atom C(12) is 2.522(10) Å. The O(2)⋯C(H)O angle is 111.9(5)° similar to the “Bürgi–Dunitz” angle and consistent with the alignment of electron density from oxygen with the anti-bonding pi orbital of the carbonyl group. The shortest contact from the aldehyde's electron deficient hydrogen atom to the carboxylate group is to oxygen O(2) (2.32 Å) and this hydrogen lies 2.55 Å from the carboxylate carbon atom. Selected molecular geometry for the anion is given in Table 1. Both carboxylate oxygen atoms coordinate the same potassium ion, as well as each making a coordination to a second potassium ion. The aldehyde oxygen atom coordinates two potassium ions – one coordination roughly aligned with an oxygen sp2 lone pair (2.778(7) Å) and the other, rather longer, approximately perpendicular to the plane of the aldehyde group (3.341(7) Å). The water molecule bridges two potassium ions (O⋯K: 2.760(7) and 2.842(7) Å) and is involved in hydrogen bonds to carboxylate oxygen atoms (O(4)–H(41)⋯O(1): 2.09 Å, angle at H(41) 162.1° and O(4)–H(42)⋯O(2): 1.94 Å, angle at H(42) 166.9°).
 |
| | Fig. 2 Crystal structure of K·14 monohydrate. | |
 |
| | Fig. 3 (a) The structure of anion 14 in its hydrated potassium salt with associated potassium ions and water molecules; (b) the structure of anion 14 showing the interaction between the two functional groups. | |
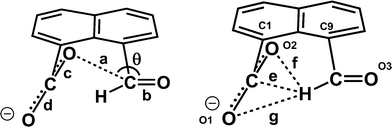
Table 1 Selected molecular geometry for the naphthalaldehydate anion 14 in salts with lithium, potassium, cesium and triphenylguanidinium cationsa
|
|
|
|
a/Å |
b/Å |
c, d/Å |
e/Å |
f, g/Å |
θ/° |
Angle between ringb/CO2− planes/° |
Angle between ringb/CHO planes/° |
Angle between CO2−/CHO planes/° |
ΔC(O2−) from ring plane/Å |
ΔC(HO) from ring plane/Å |
Splaying of groups at C1 & C9 |
|
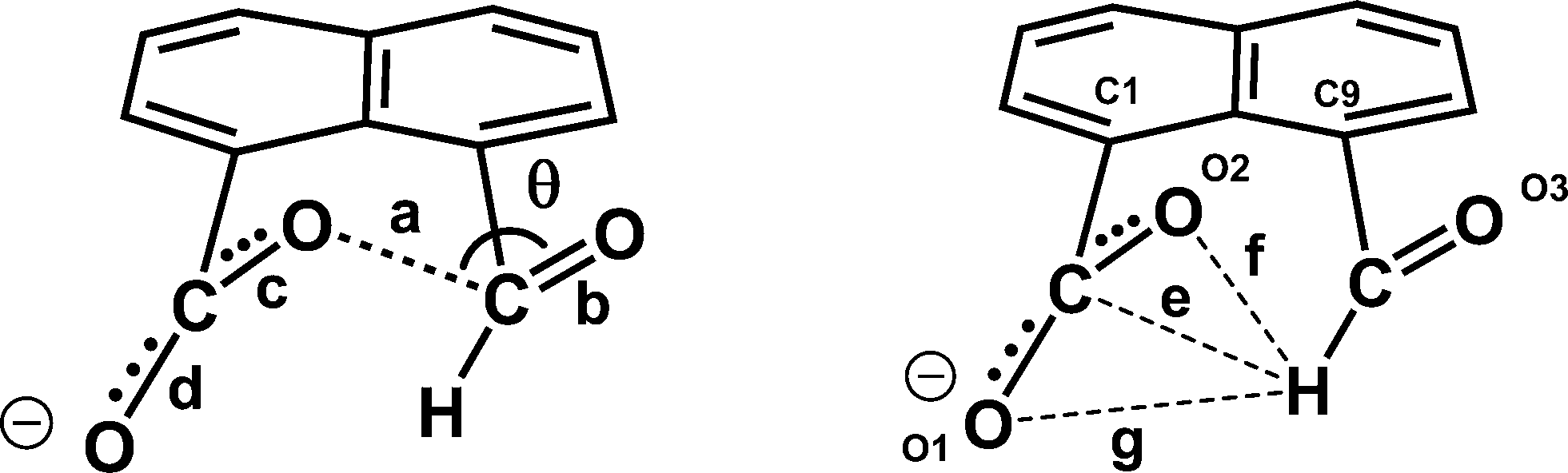
a = O(2)⋯C(12); b = O(3)–C(12); c = O(2)–C(11); d = O(1)–C(11); e = C(11)⋯H(12); f = O(2)⋯H(12); g = O(1)⋯H(12); θ = O(2)⋯C(12)–O(3).
Ring plane defined by its ten C atoms.
|
| Li·14 |
2.445(15) |
1.220(15) |
1.261(14), 1.253(13) |
2.57 |
2.33, 3.42 |
107.4(8) |
32.0(8) |
32.7(6) |
36(1) |
−0.461(16) |
0.383(16) |
−2.1, 3.8 |
| K·14 |
2.522(10) |
1.212(10) |
1.280(9), 1.241(10) |
2.55 |
2.32, 3.34 |
111.9(5) |
33.0(8) |
29.0(6) |
32(1) |
−0.368(12) |
0.425(13) |
−3.8, 4.6 |
| Cs·14 |
2.502(12) |
1.216(13) |
1.270(13), 1.248(12) |
2.56 |
2.31, 3.38 |
110.9(7) |
30.6(12) |
29.4(8) |
32(2) |
−0.408(17) |
0.439(16) |
−2.0, 3.5 |
| (PhNH)3C·14 |
2.6303(14) |
1.2115(15) |
1.2768(14), 1.2455(14) |
2.40 |
2.44, 2.92 |
111.17(8) |
47.6(1) |
28.5(1) |
31(2) |
−0.3533(16) |
0.3283(17) |
−1.9, 3.5 |
The cesium salt shows the same coordination pattern as for the potassium salt and the same asymmetry in the carboxylate group (1.248(12) and 1.270(13) Å). The carboxylate and aldehyde groups adopt similar positions with an O⋯C interaction distance of 2.502(12) Å, and a similar angle of approach to the carbonyl group (110.9(7)°) (Table 1). The aldehyde's hydrogen atom lies 2.31 Å from the carboxylate's oxygen O(2) and 2.56 Å from the carboxylate carbon atom. The eight cesium–oxygen coordination bonds lie in the range 3.052(8)–3.391(8) Å.
The crystals of the lithium salt of anion 14 crystallise in the monoclinic crystal system with space group P21/c and are twinned. The structure was refined with two twin components related by a 2-fold axis rotation about the [100] reciprocal lattice direction. The atomic connectivity is confirmed, but due to poor data quality and twinning the atomic positions and derived geometry are not very accurate.
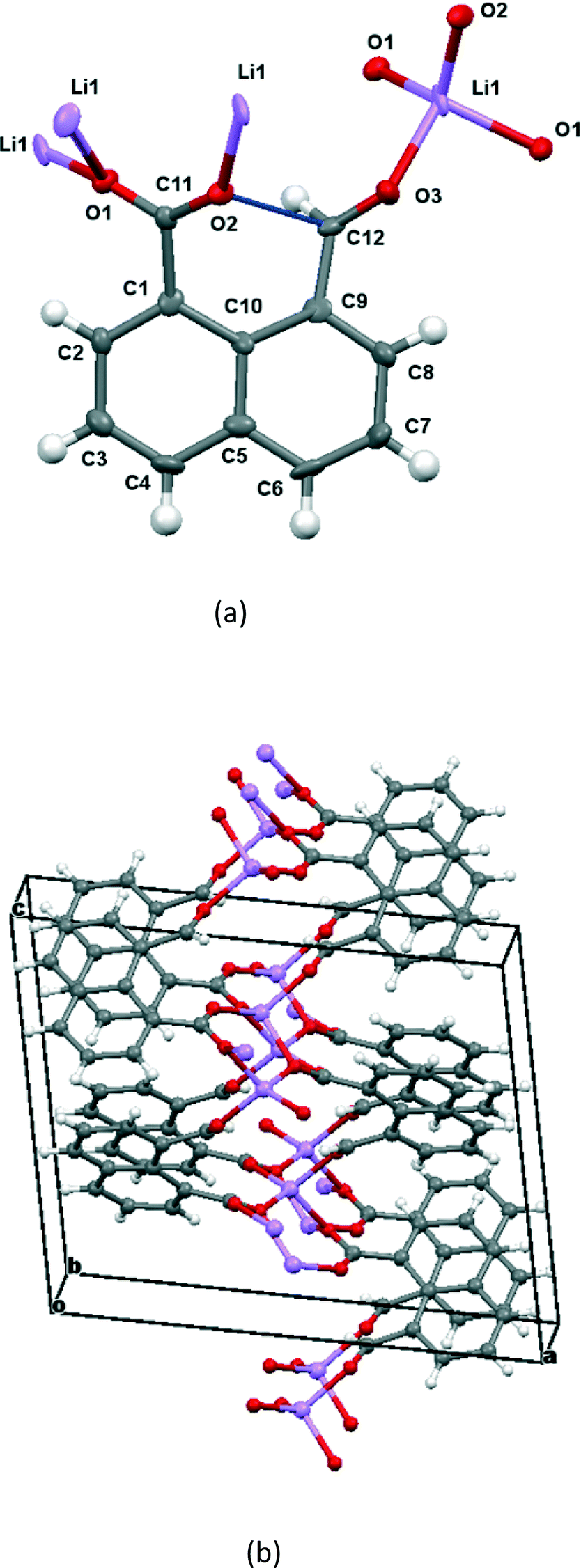
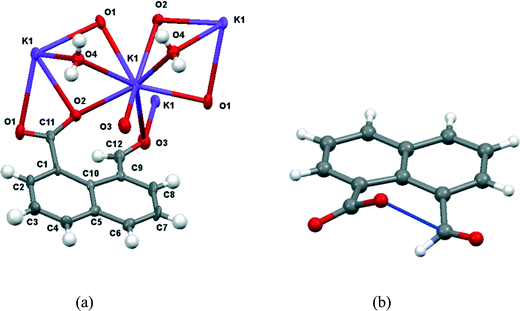
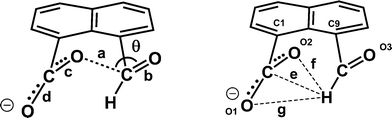
The molecular structure and crystal packing for the lithium salt are shown in Fig. 4. The lithium ions are four-coordinate and there is no water contained in the crystal lattice. The conformation of the anion is similar to that in the potassium and cesium salts, and the carboxylate/aldehyde interaction has a O(2)⋯C(12) contact of 2.445(15) Å, and a O(2)⋯C(12)O(3) angle of 107.4(8)° (Table 1). The aldehyde hydrogen lies 2.33 Å from carboxylate oxygen O(2), and 2.57 Å from the carboxylate carbon, similar to the potassium and cesium salts. The bond lengths of the carboxylate group are very similar (1.253(13) and 1.261(14) Å). The carboxylate oxygen atom remote from the aldehyde, O(1), coordinates to two lithium ions (O(1)–Li(1): 1.91(2) and 1.97(2) Å), while the second oxygen, O(2), coordinates just to one (O(2)–Li(1): 1.87(2) Å) as does the aldehyde oxygen (O(3)–Li(1): 1.95(2) Å). The angles at lithium between oxygen ligands lie in the range 97.9(10)–119.7(11)°.
 |
| | Fig. 4 (a) The molecular structure of the naphthalaldehydate anion in Li·14, showing the interaction between the two functional groups and the coordinations to the lithium ions; (b) the crystal packing arrangement of Li·14. | |
Salts with organic cations
Since both functional groups in the naphthalaldehydate anion are coordinated to metal ions which might exert an over-riding influence on the conformation of the anion, several salts with organic cations were prepared. Naphthalaldehydic acid solutions treated with sym-triphenylguanidine, N,N,N′,N′-tetramethylguanidine or 4-dimethylaminopyridine (DMAP) were left to slowly evaporate. The two guanidines gave 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salts with the naphthalaldehydic acid, while the reaction with 4-dimethylaminopyridine gave a 1:1 molecular compound between the expected DMAP salt of anion 14 and the cyclic form of naphthalaldehydic acid 15. The X-ray crystal structures of these materials were measured at low temperature. Secondary amines such as morpholine react and substitute the OH group of 15 rather than form a salt.19
:1 salts with the naphthalaldehydic acid, while the reaction with 4-dimethylaminopyridine gave a 1:1 molecular compound between the expected DMAP salt of anion 14 and the cyclic form of naphthalaldehydic acid 15. The X-ray crystal structures of these materials were measured at low temperature. Secondary amines such as morpholine react and substitute the OH group of 15 rather than form a salt.19
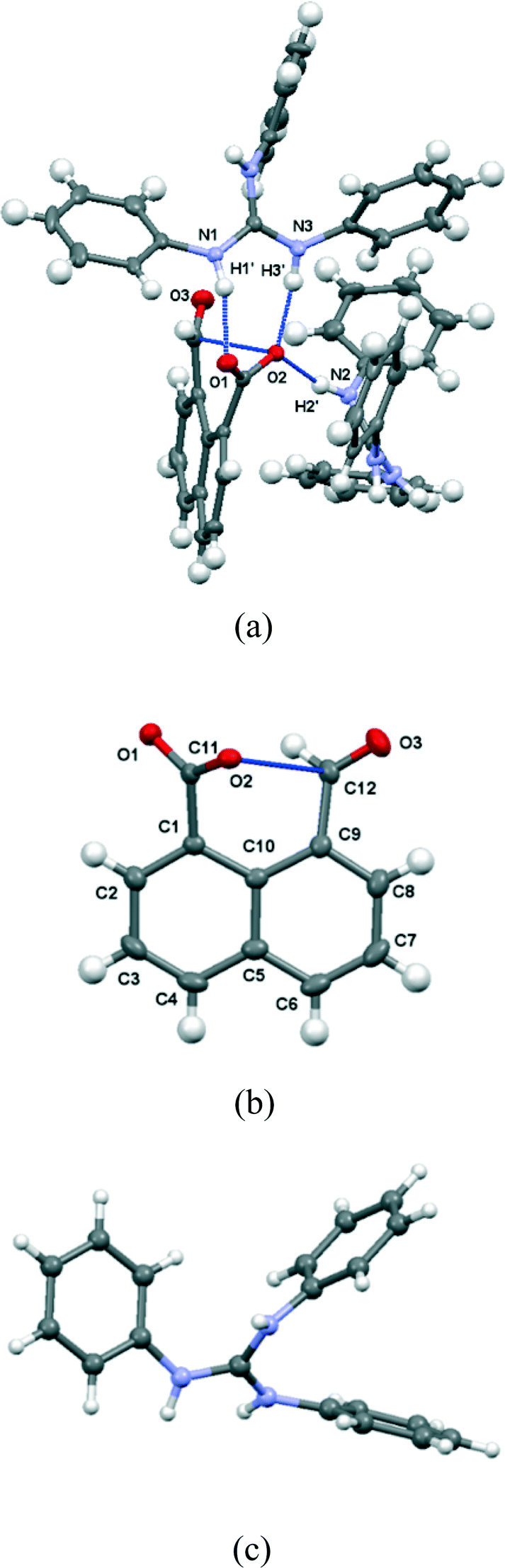
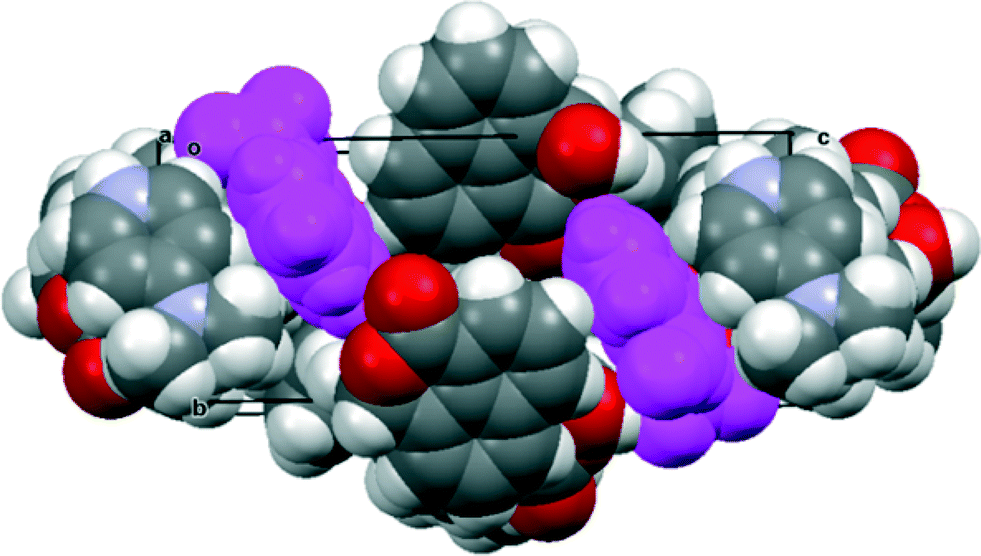
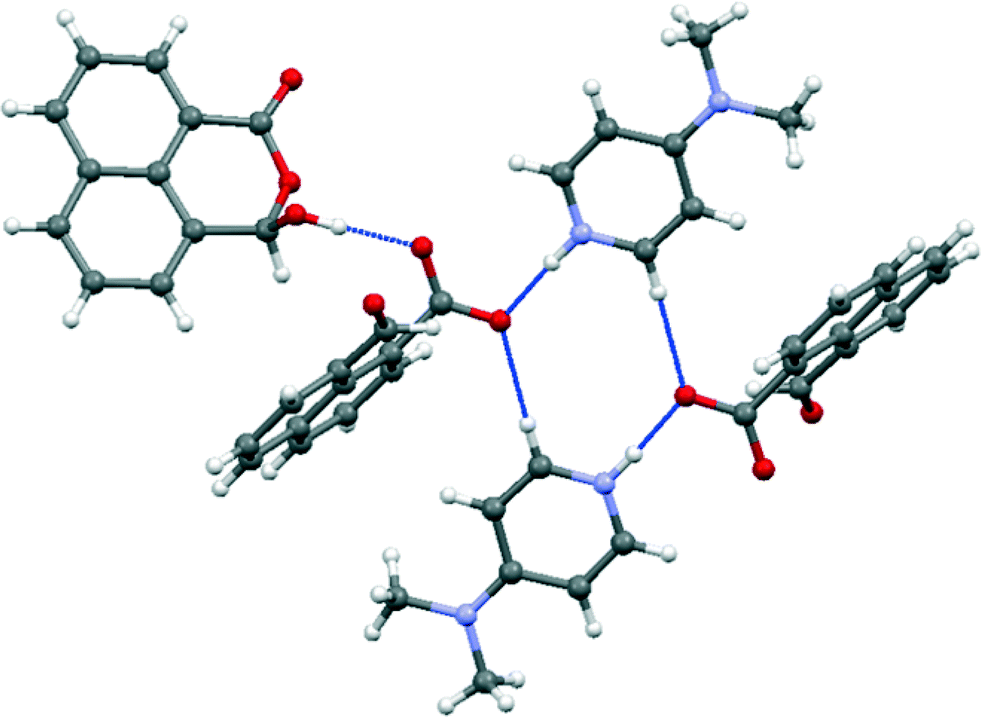
The salt with sym-triphenylguanidine crystallises from methanol in the orthorhombic crystal system in space group Pbca. The molecular structures of the cation and anion and the hydrogen bonding between them are shown in Fig. 5, and their crystal packing arrangement in Fig. 6. The triphenylguanidinium cation, rather than adopting a conformation with three fold symmetry, aligns two of its N–H bonds towards the anion. The torsion between these two N–H bonds is 44.7°, and the two hydrogen atoms make hydrogen bonds to the two oxygen atoms of the anion's carboxylate group: H(1′)⋯O(1): 1.87 Å, H(3′)⋯O(2): 1.85 Å with angles of 157.3° and 166.9° at H(1′) and H(3′) respectively. This is a well-known motif between carboxylate and guanidinium cations.20 There is a further hydrogen bond between the anion from carboxylate oxygen atom O(2) and the third guanidinium (N)–H atom from a second cation: H(2′)⋯O(2): 1.89 Å, with an angle of 162.2° at hydrogen. The anion's conformation is similar to that observed in its group 1A metal salts, and the carboxylate oxygen O(2) lies at 2.6303(14) Å from the aldehyde carbon atom, and the O(2)⋯C(12)O(3) angle is 111.17(8)° (Table 1). Thus, the carboxylate oxygen atom O(2) is involved in two hydrogen bonds and a O⋯C interaction, and the vectors of these three features lie roughly mutually perpendicular, with the hydrogen bonds lying closer to the axes of the oxygen atoms two sp2 lone pairs. The O(2)⋯C(12) interaction is slightly longer than in the metal salts, due to the greater rotation of the carboxylate anion out of the naphthalene plane presumably to optimise the formation of hydrogen bonds. The distance of the aldehyde hydrogen to the nearer carboxylate oxygen is also longer than in the group 1A salts (2.44 v. 2.31–2.33 Å), and its contact to the carboxylate carbon atom is shorter (2.40 v. 2.55–2.57 Å). 1H and 13C NMR spectra of the salt in acetone suggests that it has converted to a 1:1 mixture of triphenylguanidine and naphthalaldehydic acid in solution.
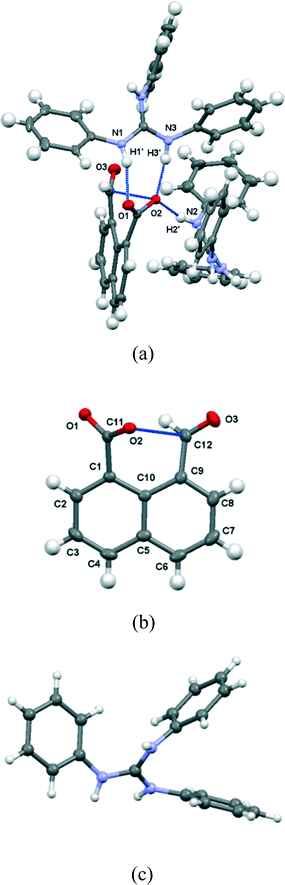 |
| | Fig. 5 (a) The interactions between the naphthalaldehydate anion 14 with two triphenylguanidinium cations in (PhNH)3C·14; (b) the structure of a single anion of 14 in (PhNH)3C·14; (c) the conformation of the cation in the crystal structure of (PhNH)3C·14; the cation is refined as two very similar conformations, only one of which is shown, differing slightly in the positions of two of the phenyl groups. | |
 |
| | Fig. 6 Crystal packing arrangement (viewed along the c-axis) for (PhNH)3C·14 with the cations shown in blue. The anions are packed in face to face pairs. | |
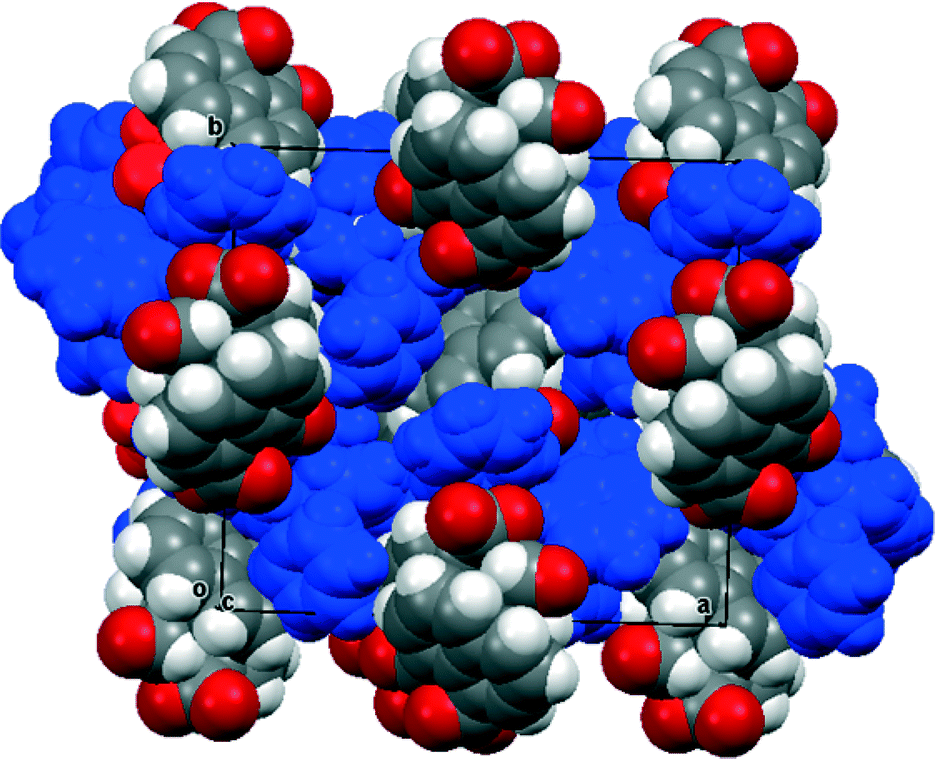
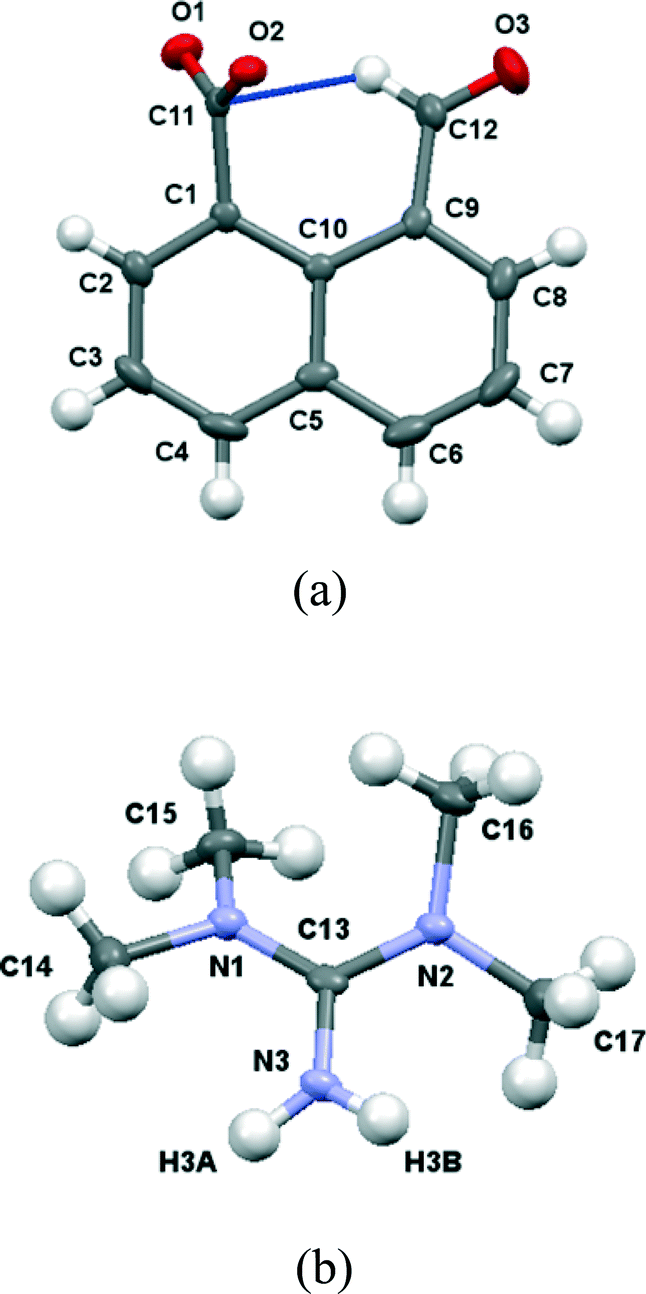
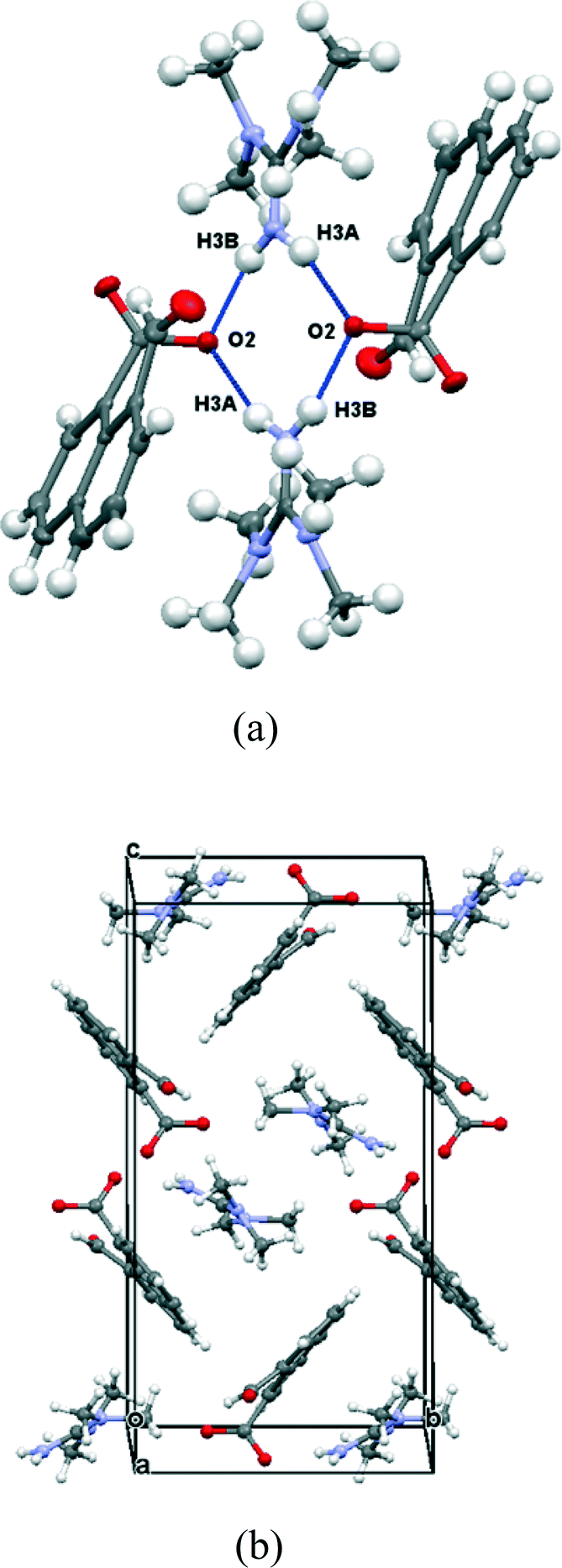
In contrast the crystal structures of the remaining two naphthalaldehydate salts showed a quite different interaction between the peri functional groups, in which the carboxylate group is strongly twisted out of the naphthalene plane and the aldehyde group directs its hydrogen atom in towards the carboxylate group. In the refinements of these two crystal structures, the positions and isotropic displacement parameters of the aldehyde's hydrogen atom and the hydrogens attached to nitrogen in the cation were refined, and not just placed in calculated positions. For the salt with N,N,N′N′-tetramethylguanidine measured at 110 K the structures of the anion and cation are shown in Fig. 7 and the hydrogen bonding arrangement and crystal packing are shown in Fig. 8. The crystal system is monoclinic and the space group P21/n. The plane of the aldehyde group lies at 21(2)° to the best plane of the naphthalene system, with the hydrogen atom directed towards the carboxylate group whose plane lies at 68.71(14) to the naphthalene system. The planes of the two functional groups lie at 58.8(5)°. There is a very short contact between the groups, from the electron deficient aldehyde hydrogen atom to the carbon of the carboxylate anion, (2.29(2) Å), and this hydrogen atom is at somewhat longer distances from both carboxylate oxygen atoms (O(1)⋯H(12): 2.54(2), O(2)⋯H(12): 2.65(2) Å). This interaction may be best viewed as an interaction of the electron deficient aldehyde hydrogen with the face of the carboxylate anion, with the hydrogen atom positioned at similar distances to the carboxylates oxygens which carry more of the net negative charge. The two functional groups are splayed apart both in the aromatic plane and perpendicular to it (Table 2). In the crystal packing arrangement hydrogen bonds connect two cations and two anions together in a “square” motif involving both hydrogens of each cation's amino group and the O(2) atom of the carboxylate group of each anion (H⋯O: 1.92(2) and 1.92(2) Å). The carboxylate group is asymmetric with the longer bond to the doubly hydrogen bonded oxygen atom (1.263(2) vs. 1.239(2) Å). The tetrasubstituted guanidinium cation is twisted to reduce steric interactions between two methyl groups, so that the N1–C15 and N2–C16 bonds make a torsion of 55.2°. The steric pressure is reduced by small pyramidalisations of the two nitrogen atom geometries, where the sums of bond angles are 358°, and the widening of bond angles C15–N1–C13 and C16–N2–C13 to 122.4–122.6° with consequent contraction of the CH3–N–CH3 angles to 114.7–115.2°.
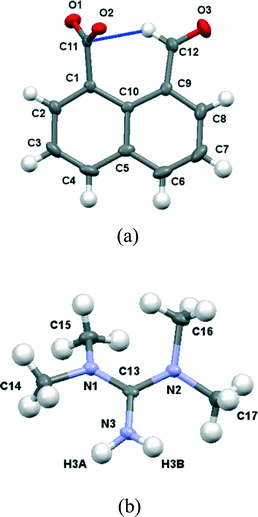 |
| | Fig. 7 (a) The structure of the naphthaldehydate anion in (Me2N)2CNH2·14 showing the interaction between the two functional groups; (b) the structure of the cation in (Me2N)2CNH2·14. | |
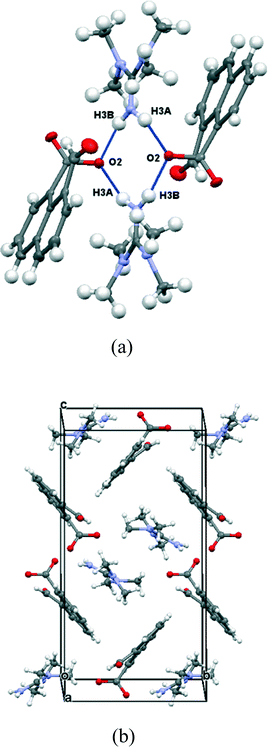 |
| | Fig. 8 (a) Hydrogen bonding linking the anions and cations in (Me2N)2CNH2·14; (b) crystal packing arrangement viewed down the a axis in (Me2N)2CNH2·14. | |
Table 2 Selected molecular geometry for the naphthalaldehydate anion 14 in salts with N,N,N′,N′-tetramethylguanidine (TMG) and 4-dimethylaminopyridine (DMAP)a
|
|
|
|
a/Å |
b/Å |
c, d/Å |
e/Å |
f, g/Å |
θ/° |
Angles c/e, d/e/° |
Angle between ringb/CO2− planes/° |
Angle between ringb/CHO planes/° |
Angle between CO2−/CHO planes/° |
ΔC(O2−) from ring plane/Å |
ΔC(HO) from ring plane/Å |
Splaying of groups at C1 & C9 |
|
a = O(2)⋯C(12); b = O(3)–C(12); c = O(2)–C(11); d = O(1)–C(11); e = C(11)⋯H(12); f = O(2)⋯H(12); g = O(1)⋯H(12); θ = O(2)⋯C(12)–O(3).
Ring plane defined by its ten C atoms.
|
|
TMG-H·14
|
3.043(2) |
1.211(2) |
1.263(2) 1.239(2) |
2.29(2) |
2.65(2), 2.54(2) |
125.64(14) |
91.7(5), 86.4(5) |
68.71(14) |
21(2) |
58.8(5) |
−0.265(2) |
0.319(3) |
−3.1, 3.6 |
| DMAP-H·14·15 |
2.920(3) |
1.184(2) |
1.240(2) 1.260(2) |
2.42(2) |
2.60(2), 2.76(2) |
119.77(19) |
83.8(6), 91.8(6) |
68.8(2) |
32(2) |
45.6(5) |
−0.065(2) |
0.106(3) |
−4.0, 4.2 |
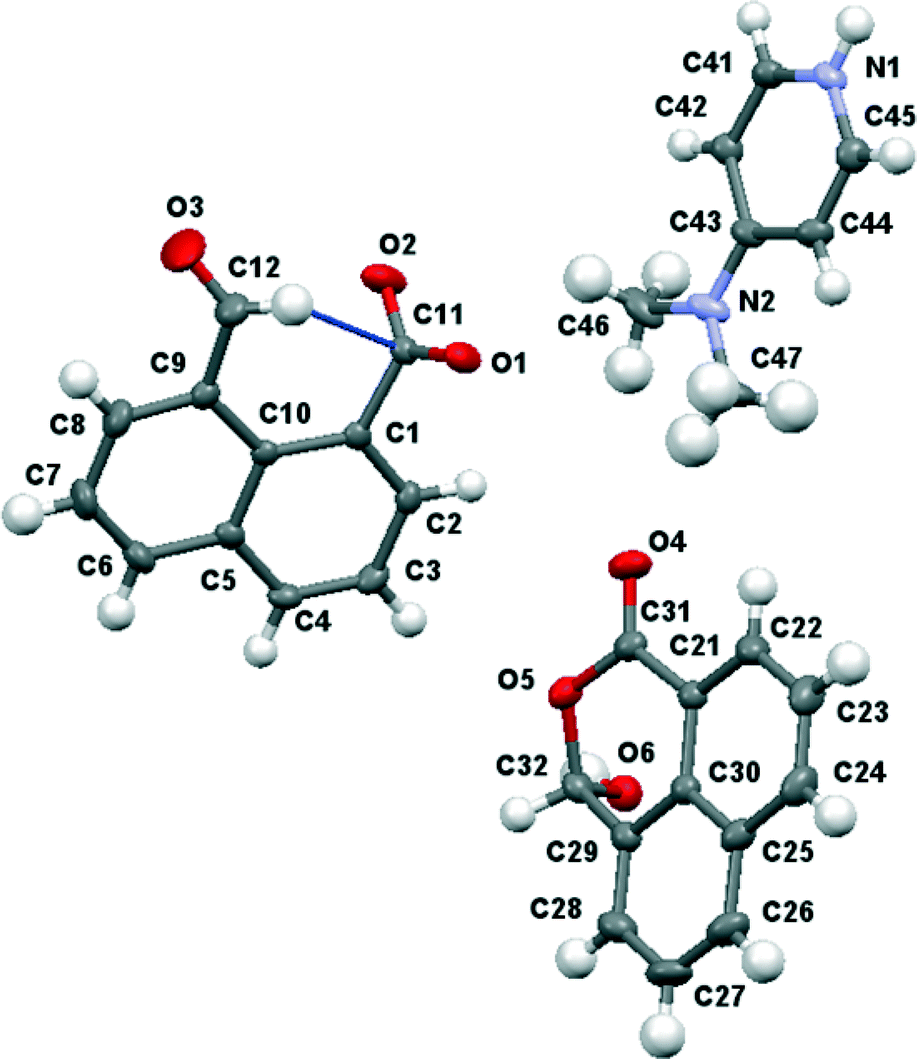
X-ray crystallography showed the material isolated by treatment of naphthalaldehydic acid with DMAP contained the 4-dimethylaminopyridium salt of naphthalaldehydic acid along with one equivalent of the acid itself in its cyclic form 15 (Fig. 9). The crystals are monoclinic with space group P21/n, and contain stacks along the a axis comprising centrosymmetric pairs of cyclised naphthaldehydic acid molecules 15 alternating with centrosymmetric pairs of 4-dimethylaminopyridinium cations. The naphthalaldehydate anions lie between the stacks with their naphthalene planes perpendicular to the other species (Fig. 10). The molecules are linked together by hydrogen bonding, thus two naphthalaldehydate anions and two dimethylaminopyridinium species are linked into a “hexagonal” arrangement by one of the carboxylate oxygens, O(1), which makes a short hydrogen bond to the N–H group (1.73 Å, O(1)⋯H(1)–N(1): 175.8°) and a rather longer one to the hydrogen at the 2-position on the pyridinium ring (2.36 Å, O(1)⋯H(41)–C(41): 175.4°). The second carboxylate oxygen atom, O(2), is hydrogen bonded to the hydroxyl group of the cyclised naphthalaldehydic acid molecule (O(2)⋯H: 1.76 Å, O(2)⋯H(61)–O(6): 178.5°) (Fig. 11). The lengths of the two carboxylate C–O bonds in anion 14 are distinctly different (O(1)–C(11): 1.260(2), O(2)–C(11): 1.240(2) Å), the longer bond being involved in two hydrogen bonds rather than one.
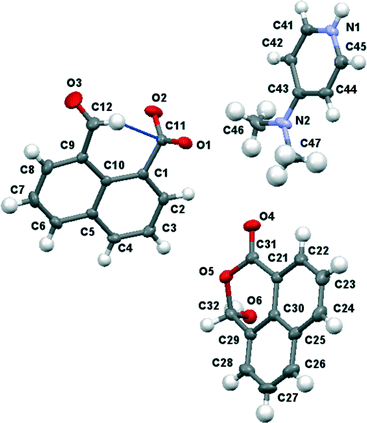 |
| | Fig. 9 Molecular structures of the three components of the co-crystal between 4-dimethylaminopyridinium naphthalaldehydate and naphthalaldehydic acid. | |
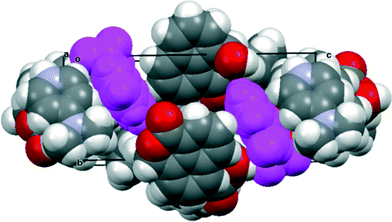 |
| | Fig. 10 Crystal packing of the co-crystal between 4-dimethylpyridinium naphthalaldehydate and naphthalaldehydic acid, viewed down the a axis with the naphthalaldehydate anions shown in magenta. AA′BB′ stacks of the other components lie along the a axis. | |
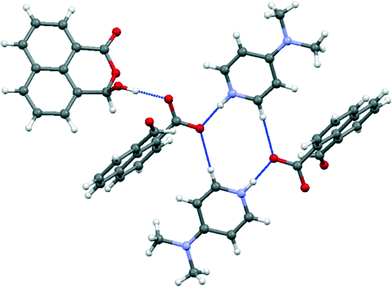 |
| | Fig. 11 Hydrogen bonding arrangement in the co-crystal between 4-dimethylpyridinium naphthalaldehydate and naphthalaldehydic acid. | |
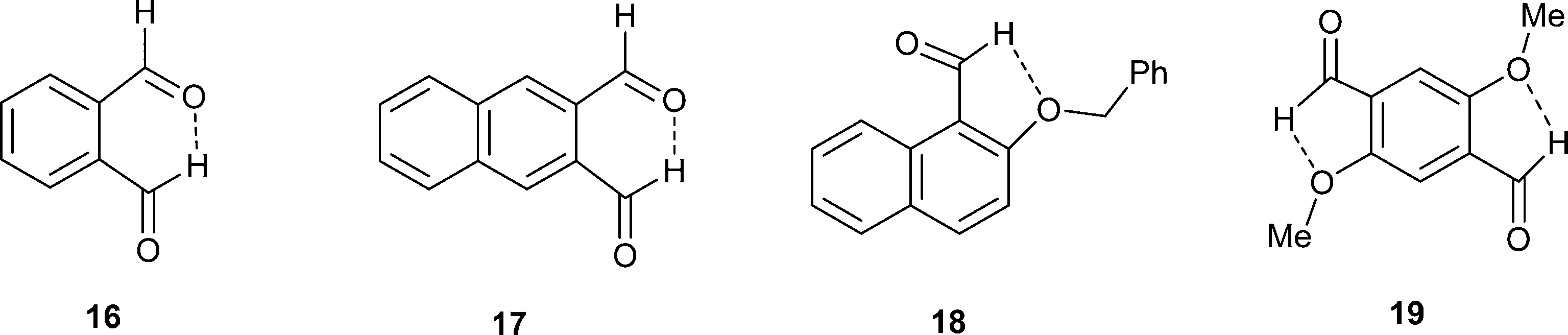
In the anion, the aldehyde and carboxylate groups lie at 45.6(5)°, a smaller angle than in the tetramethylguanidium salt (60.5(2)°). The aldehyde hydrogen is directed towards the carboxylate carbon with a H⋯CO2− separation of 2.42(2) Å, and distances of 2.76(2) and 2.60(2) Å to the two oxygen atoms. The smaller angle between the functional group planes means that there is also a rather long contact from a carboxylate oxygen to the aldehyde carbon (O(2)⋯CHO: 2.920(3) Å) with a O(2)⋯CO angle of 119.77(19)°. The structures of the anion in the salts with tetramethylguanidine and DMAP are compared in Fig. 12 along with those for the potassium and triphenylguanidinium salts. The greater angle between the functional group planes in the tetramethylguanidium salt means that the H⋯CO2− contact is shorter (2.29(2) Å), and the O⋯CHO contact longer (3.043(2) Å). There are no similar short contacts between an aldehyde and the carbon of a carboxylate group reported in the Cambridge Structural Database (2015 release). In the structures of the two reported salts of the 2-formylbenzoate anion the main interaction between the functional groups is an intramolecular H⋯O contact (2.26 and 2.47 Å) between the aldehyde hydrogen and a carboxylate oxygen.21 Furthermore, there are short intramolecular H⋯O contacts between aldehyde groups in benzene-1,2-dicarbaldehyde 16 and naphthalene-2,3-dicarbaldehyde 17 (2.30–2.36 Å), but in benzene-1,3-dicarbaldehyde the intermolecular H⋯O contacts involving aldehyde groups are longer (2.64–2.66 Å).22 Two similar contacts with an ester carbonyl oxygen (2.43 Å) have been reported.23 The aldehyde group is known to make short contacts from the hydrogen atom to an ether oxygen of ca. 2.3 Å, for example in 18–19 (Scheme 4).24
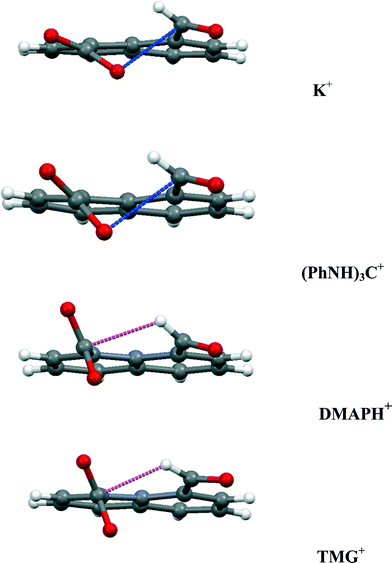 |
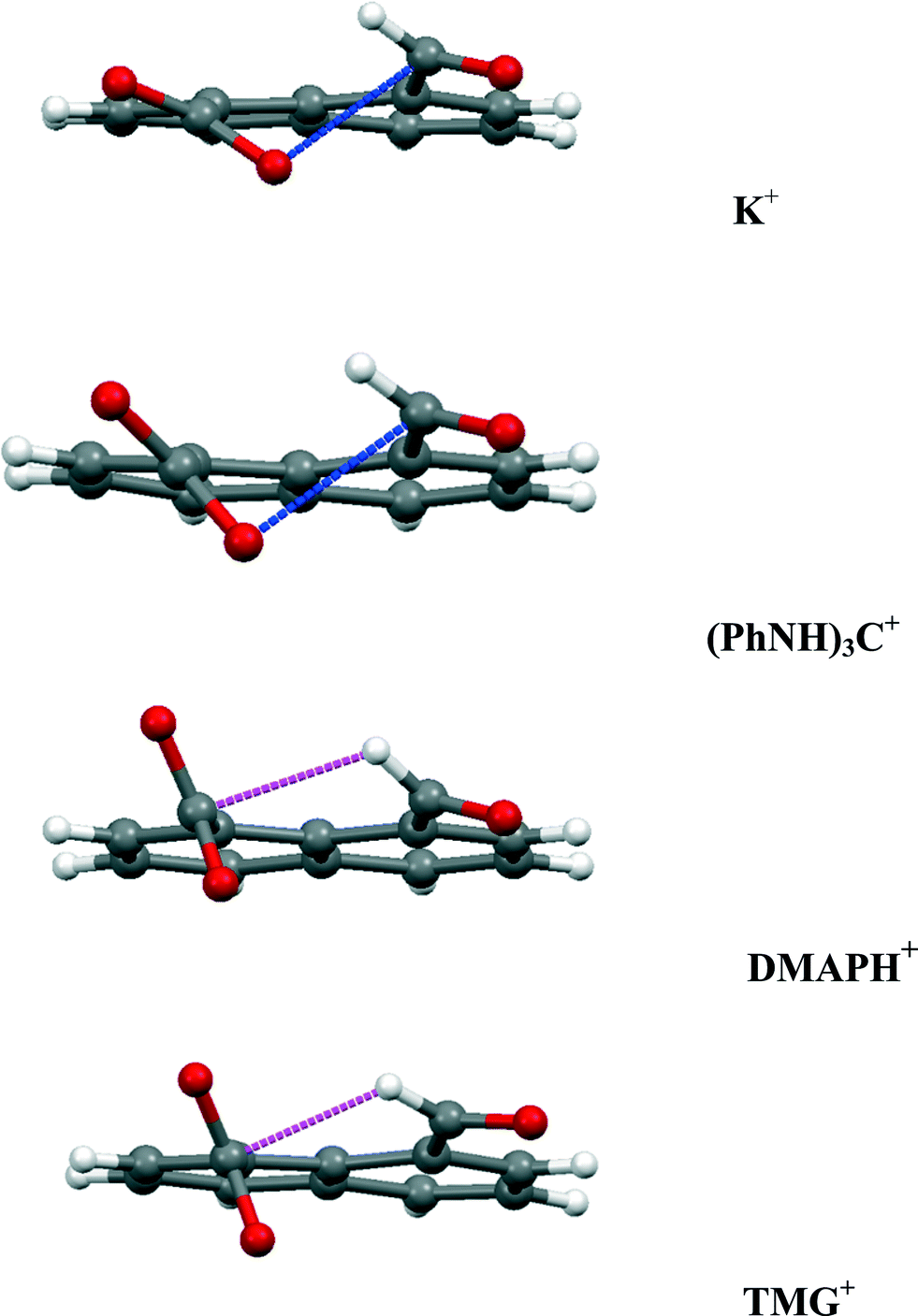
| | Fig. 12 Comparison of the interactions between the functional groups in the anion 14 for four of its salts, showing just the dominant interaction. The O⋯C distance (blue) increases down the series from 2.522 to 3.043 Å, and the H⋯CO2− (magenta) distance decreases from 2.55 to 2.29 Å down the series. | |
 |
| | Scheme 4 Hydrogen bonding from oxygen to the hydrogen atom of an aldehyde group. | |
Conclusion
Two types of interaction between the carboxylate and aldehyde groups have been observed in a peri-naphthalene system. In one mode a negatively charged oxygen impinges on an aldehyde carbonyl for which there are only a few examples, such as in salts of aspirin with organic cations but for which the O⋯C contacts are longer (>2.75 Å).25 Compared to the peri-interactions between methoxy groups and various electrophilic groups, the use of a negatively charged oxygen as the nucleophile has led to slightly shorter O⋯C interactions down to 2.45 Å while maintaining the angle of approach of the oxygen to the carbonyl group in the 107–112° range. This is achieved by in-plane and out of plane splayings of the groups. In the neutral parent acid a covalent O–C bond is formed between the two groups (1.477(2) Å). In the second mode the aldehyde has directed its hydrogen atom at the face of the carboxylate anion so that it makes a particularly short contact with the carbon atom (2.29–2.42 Å), for which there is no example in the Cambridge Structural Database. The interactions in four of the salts are compared in Fig. 12. As the carboxylate group rotates further out of the aromatic plane, the O⋯CO contact gets longer, and the H⋯CO2− contact gets shorter. These interaction modes will be studied further using cations which make the minimal interaction with the anion, and the interactions investigated in more detail by conducting charge density measurements.
Experimental
General
Solution NMR spectra were measured on a Jeol ECLIPSE 400 spectrometer at 400 MHz for 1H and at 100.6 MHz for 13C using CDCl3 as solvent and tetramethylsilane (TMS) as standard unless otherwise stated, and measured in p.p.m. downfield from TMS with coupling constants reported in Hz. IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR Spectrometer using Attenuated Total Reflection sampling unless otherwise stated, and are reported in cm−1. Chemical analysis data were obtained from Mr. Stephen Boyer, London Metropolitan University.
1,8-Naphthalaldehydic acid 13
The material was purchased from Sigma-Aldrich. δH (400 MHz, ((CD3)2CO, 24 °C): 8.37 (1H, dd, J = 7.2, 1.2 Hz, Ar–H1), 8.33 (1H, dd, J = 8.3, 1.1 Hz, Ar–H1), 8.11 (1H, dd, J = 8.1, 1.2 Hz, Ar–H1), 7.70–7.81 (3H, m, Ar–H3), 7.16 (1H, s, OH), 6.94 (1H, s, CHOH); δc (75.5 MHz, (CD3)2CO, 24 °C): 163.7 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O), 134.5, 133.0, 130.9 130.0, 129.1, 128.1, 127.7, 127.2, 126.1, 121.3 (Ar–C10), 97.1 (CHOH); δH (400 MHz, CD3OD, 24 °C): 8.32 (1H, d, J = 7.3 Hz, Ar–H1), 8.19 (1H, d, J = 8.7 Hz, Ar–H1), 7.98 (1H, d, J = 7.8 Hz, Ar–H1), 7.63–7.68 (3H, m, Ar–H3), 7.16 (1H, s, OH), 6.81 (1H, s, CHOH); δc (100.6 MHz, CD3OD, 24 °C): 165.9 (CO), 135.2, 133.4, 130.8 130.5, 129.5, 128.4, 128.0, 127.4, 126.5, 120.9 (Ar–C10), 99.0 (CHOH); νmax: 3274, 1675, 1590, 1587, 1517, 1407, 1377, 1356, 1305, 1264, 1249, 1181, 1113, 992, 922, 771, 740, 687, 655 cm−1. Data are consistent with cyclic structure 15.
O), 134.5, 133.0, 130.9 130.0, 129.1, 128.1, 127.7, 127.2, 126.1, 121.3 (Ar–C10), 97.1 (CHOH); δH (400 MHz, CD3OD, 24 °C): 8.32 (1H, d, J = 7.3 Hz, Ar–H1), 8.19 (1H, d, J = 8.7 Hz, Ar–H1), 7.98 (1H, d, J = 7.8 Hz, Ar–H1), 7.63–7.68 (3H, m, Ar–H3), 7.16 (1H, s, OH), 6.81 (1H, s, CHOH); δc (100.6 MHz, CD3OD, 24 °C): 165.9 (CO), 135.2, 133.4, 130.8 130.5, 129.5, 128.4, 128.0, 127.4, 126.5, 120.9 (Ar–C10), 99.0 (CHOH); νmax: 3274, 1675, 1590, 1587, 1517, 1407, 1377, 1356, 1305, 1264, 1249, 1181, 1113, 992, 922, 771, 740, 687, 655 cm−1. Data are consistent with cyclic structure 15.
Preparation of the lithium salt of 13
A solution of LiOH·H2O (0.11 g, 2.62 mmol) in water (2 ml) was added to a solution of 1,8-naphthalaldehydic acid 13 (0.50 g, 2.49 mmol) in hot methanol (6 ml), and the mixture stirred and then left to cool. Beige crystals were filtered off and washed with ethanol and ether to give lithium naphthalaldehydate (0.39 g, 75%), m.p. 327–329 °C. δH (400 MHz, D2O, 24 °C): 10.14 (1H, s, CHO), 8.09 (1H, dd, J = 8.2, 1.1 Hz, Ar–H1), 7.98 (1H, dd, J = 8.3, 1.2 Hz, Ar–H1), 7.91 (1H, dd, J = 7.1, 1.3 Hz, Ar–H1), 7.83 (1H, dd, J = 7.2, 1.3 Hz, Ar–H1), 7.59 (1H, br t, J = 7.7 Hz, Ar–H1), 7.57 (1H, br t, J = 7.8 Hz, Ar–H1); δc (75.5 MHz, D2O, 24 °C): 196.4 (CHO), 180.8 (CO2−), 137.9, 137.6, 136.6, 135.7, 133.2, 133.0, 130.8, 129.4, 128.9, 128.0 (Ar–C10); νmax: 3050, 2875, 1674, 1617, 1582, 1564, 1503, 1426, 1415, 1390, 1352, 1342, 1244, 1205, 1172, 1150, 1105, 1088, 1012, 963, 926, 851, 830, 794, 782, 755, 692 cm−1. Found C: 70.03 H: 3.34%. C12H7O3Li requires C: 69.92 H: 3.42%.
Preparation of the potassium salt of 13
Naphthalaldehydic acid (0.50 g, 2.49 mmol) 13 was added to an aqueous solution of potassium hydroxide (2 ml, 1.25 M, 2.50 mmol) and the mixture was stirred and then left to stand. After 1 day, the solvent was evaporated and dissolved in warm isopropanol and left to cool and evaporate. The crystals that formed were collected, and washed with isopropanol and diethyl ether to give hydrated potassium naphthalaldehydate (0.15 g, 23%) as white crystals, m.p. 290 °C dec. δH (300 MHz, CD3OD, 24 °C): 10.47 (1H, s, CHO), 8.12 (1H, dd, J = 8.2, 1.3 Hz, Ar–H1), 7.96–8.01 (2H, m, Ar–H2), 7.88 (1H, dd, J = 7. 1, 1.3 Hz, Ar–H1), 7.54–7.62 (2H, m, Ar–H2); δc (75.5 MHz, CD3OD, 24 °C): 192.7 (CHO), 178.9 (CO2−), 138.6, 135.9, 135.7, 135.2, 130.7, 129.4, 129.2, 128.6, 127.0, 126.1 (Ar–C10); νmax: 3389 br, 3053, 2896, 1677, 1619, 1596, 1578, 1559, 1426, 1407, 1375, 1340, 1240, 1217, 1203, 1167, 1102, 1081, 1015, 972, 956, 931, 845, 828, 794, 783, 759, 687 cm−1. Found C: 56.34, H: 3.64%. C12H7O3K·H2O requires C: 56.24, H: 3.54%.
Preparation of the cesium salt of 13
Naphthalaldehydic acid (0.20 g, 1.00 mmol) 13 and cesium hydroxide monohydrate (0.168 g, 1.00 mmol) were warmed in methanol (3 ml) to give a yellow solution, and then left to evaporate. The residue was recrystallized from warm ethanol containing a few drops of water to give hydrated cesium naphthalaldehydate (0.076 g, 22%) as white plates, m.p. 272–275 °C dec. δH (400 MHz, CD3OD, 24 °C): 10.50 (1H, s, CHO), 8.11 (1H, dd, J = 8.2, 1.4 Hz, Ar–H1), 7.96–8.00 (2H, m, Ar–H2), 7.88 (1H, dd, J = 7.3, 1.4 Hz, Ar–H1), 7.54–7.61 (2H, m, Ar–H2); δc (100.5 MHz, CD3OD, 24 °C): 192.2 (CHO), 178.7 (CO2−), 138.5, 135.8, 135.7, 135.1, 130.8, 129.5, 129.1, 128.8, 127.0, 126.1 (Ar–C10); νmax: 3380 br, 3055, 2890, 2874, 1675, 1616, 1592, 1576, 1561, 1500, 1426, 1404, 1371, 1337, 1237, 1214, 1205, 1170, 1100, 1081, 1012, 971, 955, 923, 845, 824, 793, 781, 761, 751, 685, 622 cm−1. Found C: 41.33, H: 2.51%. C12H7O3Cs·H2O requires C: 41.17, H: 2.59%.
Reaction of 13 with 4-dimethylaminopyridine
4-Dimethylaminopyridine (0.31 g, 2.53 mmol) was added to a stirred solution of 1,8-naphthalaldehydic acid 13 (0.50 g, 2.49 mmol) in hot methanol (6 ml) and the reaction mixture was stirred for 1 h and left to cool. The volume of methanol was reduced to 1ml by evaporation and ether added and the mixture left to crystallise. The product was filtered off and washed with ether to give a molecular complex between 4-dimethylaminopyridinium naphthalaldehydate and naphthalaldehydic acid, DMAPH.14.15, (0.63 g, 96%) m.p. 143–144 °C. δH (400 MHz, (CD3)2CO, 24 °C): 8.37 (1H, dd, J = 7.2, 1.1 Hz, Ar–H1), 8.31 (1H, dd, J = 8.3, 0.9 Hz, Ar–H1), 8.08–8.11 (3H, m, 2-,6-H (DMAP), Ar–H1), 7.69–7.79 (3H, m, Ar–H3), 6.94 (1H, s, CHOH), 6.56 (2H, dd, J = 5.0, 1.6 Hz, 3-,5-H (DMAP)), 3.10 (1H, v br, OH), 2.98 (6H, s, N(CH3)2 (DMAP)); δc (75.5 MHz, (CD3)2CO, 24 °C): 163.7 (CO), 155.0 (4-C, DMAP), 150.1 (2-,6-C, DMAP), 134.4, 133.0, 131.0 129.8, 129.1, 128.1, 127.7, 127.2, 126.1, 121.3 (Ar–C10), 107.4 (3-,5-C, DMAP), 97.2 (CHOH); corresponds to 2:1 mixture of naphthaldehydic acid and DMAP; νmax: 1713, 1681, 1646 (N–H, DMAPH), 1559, 1537, 1534, 1516, 1506, 1443, 1431, 1394, 1374, 1347, 1336, 1316, 1258, 1243, 1214, 1175, 1092, 1077, 1067, 989, 955, 927, 841, 826, 810, 774, 758, 743, 732, 683, 654 cm−1; found C: 71.35, H: 4.87, N: 5.48%. C31H26O3CN2O6 requires C: 71.25, H: 5.02, N: 5.36%.
Preparation of the tetramethylguanidinium salt of 13
1,8-Naphthalaldehydic acid (0.50 g, 2.49 mmol) was dissolved in THF (5 ml) in a vial and 1,1,3,3-tetramethylguanidine (0.32 ml, 2.55 mmol) added and the vial stored in a desiccator over calcium chloride. Colourless block-like crystals (0.23 g, 29%) grew over a period of a few days. The crystals were hygroscopic, m.p. 131–133 °C; δH (400 MHz, CD3OD, 24 °C): 10.55 (1H, s, CHO), 8.10 (1H, dd, J = 8.0, 1.2 Hz, Ar–H1), 7.68–7.99 (2H, m, Ar–H2), 7.88 (1H, dd, J = 7.3, 1.4 Hz, Ar–H1), 7.54–7.59 (2H, m, Ar–H2), 5.08 (2H, br, –NH2), 2.87 (12H, s, 2 × N(CH3)2); δC (100.5 MHz, CD3OD, 24 °C): 189.9 (HCO) 177.2 (CO2−), 161.8 (N2CN), 137.3, 134.5, 134.3, 133.9, 129.5, 128.0, 127.8, 127.3, 125.7, 124.9 (Ar–C10), 39.8 (2 × N–(CH3)2); νmax: 2935 br, 1675, 1567, 1504, 1471, 1454, 1428, 1406, 1376, 1358, 1333, 1257, 1239, 1206, 1099, 1086, 1059, 1036, 954, 880, 833, 822, 802, 770, 753, 719, 681 cm−1.
Preparation of the triphenylguanidinium salt of 13
1,8-Naphthalaldehydic acid (0.20 g, 1.0 mmol) and sym-triphenylguanidine (0.29 g, 1.0 mmol) were dissolved in methanol (7 ml) and left to stand in an open flask for 36 h. As the methanol slowly evaporated, colourless crystals of triphenylguanidinium naphthalaldehydate formed, which were filtered off and washed with ether (0.107 g, 22%), m.p. 129–131 °C. The NMR in (CD3)2CO corresponded to a mixture of triphenylguanidine and naphthalaldehydic acid; νmax: 3055, 2700 br, 1686, 1659, 1640, 1596, 1586, 1536, 1493, 1460, 1450, 1375, 1335, 1273, 1237, 1222, 911, 886, 844, 799, 779, 763, 749, 731, 689, 636 cm−1; found: C: 76.47, H: 5.26, N: 8.68%, C31H25N3O3 requires C: 76.37, H: 5.17, N: 8.62%.
X-ray crystallography
Low temperature (100–150 K) X-ray diffraction data (Mo Kα) for compounds 15, K·14, Cs·14, and (PhNH)3·14 were collected at the UK National Crystallography Service,26 University of Southampton on a Rigaku AFC12 diffractometer equipped with enhanced sensitivity (HG) Saturn724+ CCD detector mounted at the window of an FR-E+ SuperBright rotating anode generator (Mo Kα, λ = 0.71075 Å) with VHF Varimax optics (70 μm focus) using Crystal Clear software27 for data collection and reduction. For compounds Li·14, DMAPH·14·15 and (MeNH)2CNH2·14 data were measured on an Agilent Xcalibur diffractometer equipped with a Sapphire detector and an 700 series Cryostream low temperature system at Nottingham Trent University using the CrysAlis-Pro software package.28 Structures were solved are refined using the SHELXS and SHELXL suite of programs29 using the XSEED interface.30 Molecular illustrations were made with Mercury.31 Data are deposited at the Cambridge Crystallographic Data Centre with code numbers CCDC 1435389–1435396.
Crystal data for 15.
C12H8O3, Mr = 200.18, monoclinic, a = 12.135(15), b = 3.979(5), c = 18.38(2) Å, β = 91.40(3)°, V = 887.2(18) Å3, Z = 4, P21/c, Dc = 1.50 g cm−3, μ = 0.108 mm−1, T = 100(2) K, 2007 unique reflections (Rint = 0.037), 1431 with F2 > 2σ, R(F, F2 > 2σ) = 0.043, Rw (F2, all data) = 0.112. Crystals grown from acetonitrile. The hydroxyl hydrogen atom was allowed to refine without restraints.
Crystal data for Li·14.
C12H7O3Li, Mr = 206.12, monoclinic, a = 14.953(2), b = 5.0185(6), c = 12.3691(19) Å, β = 100.389(13)°, V = 913.0(2) Å3, Z = 4, P21/c, Dc = 1.50 g cm−3, μ = 0.106 mm−1, T = 151(2) K, 1601 unique reflections, 1438 with F2 > 2σ, R(F, F2 > 2σ) = 0.163, Rw (F2, all data) = 0.472. Crystals grown from methanol/water. The structure was refined as a 43:57 mixture of two twin components with twin matrix [1 0 0.44 0 −1 0 0 0 −1] (2-fold axis rotation about [1 0 0] reciprocal lattice direction).
Crystal data for K·14·H2O.
C12H7O3K·H2O, Mr = 256.29, orthorhombic, a = 7.3837(4), b = 30.138(2), c = 4.8965(3) Å, V = 1089.61(11) Å3, Z = 4, Pna21, Dc = 1.56 g cm−3, μ = 0.486 mm−1, T = 123(2) K, 1855 unique reflections (Rint = 0.054), 1677 with F2 > 2σ, R(F, F2 > 2σ) = 0.081, Rw (F2, all data) = 0.188. Crystals grown from isopropanol.
Crystal data for Cs·14·H2O.
C12H7O3Cs·H2O, Mr = 350.10, orthorhombic, a = 7.852(6), b = 29.89(2), c = 4.998(3) Å, V = 1173.0(15) Å3, Z = 4, Pna21, Dc = 1.98 g cm−3, μ = 3.16 mm−1, T = 100(2) K, 2337 unique reflections (Rint = 0.058), 1979 with F2 > 2σ, R(F, F2 > 2σ) = 0.048, Rw (F2, all data) = 0.099. Crystals grown from ethanol/water.
Crystal data for DMAPH·14·15.
C7H11N2+·C12H7O3−·C12H8O3, Mr = 524.55, monoclinic, a = 15.3271(4), b = 8.3575(3), c = 20.6960(6) Å, β = 105.966(3)°, V = 2548.81(14) Å3, Z = 4, P21/n, Dc = 1.37 g cm−3, μ = 0.095 mm−1, T = 150(2) K, 6036 unique reflections (Rint = 0.035), 4920 with F2 > 2σ, R(F, F2 > 2σ) = 0.063, Rw (F2, all data) = 0.142. Crystals grown from acetonitrile.
Crystal data for (Me2N)2CNH2·14.
C5H14N3+·C12H7O3−, Mr = 315.37, monoclinic, a = 9.0149(5), b = 9.7808(5), c = 18.7439(11) Å, β = 102.981(5)°, V = 1610.47(16) Å3, Z = 4, P21/n, Dc = 1.30 g cm−3, μ = 0.091 mm−1, T = 110(2) K, 3294 unique reflections (Rint = 0.024), 2697 with F2 > 2σ, R(F, F2 > 2σ) = 0.053, Rw (F2, all data) = 0.107. Crystals were grown from THF in a desiccator since they slowly decompose on exposure to ambient conditions.
Crystal data for (PhNH)3C·14.
C12H7O3·C19H18N3, Mr = 487.54, orthorhombic, a = 20.9749(14), b = 19.1882(6), c = 13.2738(4) Å, V = 5342.3(4) Å3, Z = 8, Pbca, Dc = 1.21 g cm−3, μ = 0.079 mm−1, T = 100(2) K, 6106 unique reflections (Rint = 0.035), 4981 with F2 > 2σ, R(F, F2 > 2σ) = 0.039, Rw (F2, all data) = 0.100. Crystals grown from methanol.
Acknowledgements
We thank the EPSRC for funding the UK National Crystallography Service. We acknowledge the use of the EPSRC funded National Chemical Database Service hosted by the Royal Society of Chemistry.32 We thank Nottingham Trent University for support including diffraction facilities, and the Erasmus program for supporting GS, LNM and DH.
References
- R. W. Newberry, G. J. Bartlett, B. VanVeller, D. N. Woolfson and R. T. Raines, Protein Sci., 2014, 23, 284–288 CrossRef CAS PubMed; G. J. Bartlett, R. W. Newberry, B. VanVeller, R. T. Raines and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 18682–18688 CrossRef PubMed.
- R. W. Newberry and R. T. Raines, Chem. Commun., 2013, 49, 7699–7701 RSC.
- A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS PubMed.
- A. Choudhary, C. G. Fry, K. J. Kamer and R. T. Raines, Chem. Commun., 2013, 49, 8166–8168 RSC.
- A. Choudhary, R. W. Newberry and R. T. Raines, Org. Lett., 2014, 16, 3421–3423 CrossRef CAS PubMed.
- A. Choudhary, K. J. Kamer and R. T. Raines, J. Org. Chem., 2011, 76, 7933–7937 CrossRef CAS PubMed.
-
J. D. Dunitz, “X-ray Analysis and the Structure of Organic Molecules”, Verlag Helvetica Chimica Acta, Zürich, Switzerland, 1995, pp. 363–390 CrossRef CAS;
H. B. Burgi and J. D. Dunitz, “Structure Correlation”, VCH, Weinheim, 1994, ch. 6, vol. 1 CrossRef CAS; W. B. Schweizer, G. Procter, M. Kaftory and J. D. Dunitz, Helv. Chim. Acta, 1978, 61, 2783–2808 CrossRef CAS; D. R. W. Hodgson, N. Feeder and A. J. Kirby, J. Chem. Soc., Perkin Trans. 1, 1999, 949–954 RSC.
- P. C. Bell and J. D. Wallis, Chem. Commun., 1999, 257–258 RSC.
- J. O'Leary, W. Skranc, X. Formosa and J. D. Wallis, Org. Biomol. Chem., 2005, 3, 3273–3283 Search PubMed.
- A. Lari, M. B. Pitak, S. J. Coles, G. J. Rees, S. P. Day, M. E. Smith, J. V. Hanna and J. D. Wallis, Org. Biomol. Chem., 2012, 10, 7763–7779 CAS.
- J. O'Leary and J. D. Wallis, Org. Biomol. Chem., 2009, 7, 225–228 Search PubMed.
- E. Bresco, M. Pitak, S. J. Coles, M. Pilkington and J. D. Wallis, CrystEngComm, 2011, 13, 6978–6984 RSC.
- G. I. Birnbaum, J. Am. Chem. Soc., 1974, 96, 6165–6168 CrossRef CAS PubMed;
A. S. Cieplak, in Structure Correlation, ed. H.-B. Bürgi and J. D. Dunitz, VCH Weinheim, 1994, pp. 205–302 Search PubMed.
- A. Wannebroucq, M. B. Pitak, S. J. Coles and J. D. Wallis, Pure Appl. Chem. Search PubMed submitted.
- N. Mercadal, S. P. Day, A. Jarmyn, M. B. Pitak, S. J. Coles, C. Wilson, G. J. Rees, J. V. Hanna and J. D. Wallis, CrystEngComm, 2014, 16, 8363–8374 RSC.
- P. C. Bell, J. O'Leary, J. D. Wallis and W. B. Schweizer, J. Chem. Soc., Perkin Trans. 2, 2001, 133–139 Search PubMed.
- C. Mowbray, W. Skranc and J. D. Wallis, J. Chem. Crystallogr., 1999, 29, 335–341 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- see Supplementary material.
- J. Janczak, Cryst. Growth Des., 2015, 15, 5097–5111 CAS.
- O. Büyükgüngör and M. Odabasoglu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o2749 CAS; M. Odabasoglu and O. Büyükgüngör, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, o186 Search PubMed.
- D. Britton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o637–o639 CAS; D. Britton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 978–980 Search PubMed.
- A. Conde, A. López-Castro, R. Márquez, J. P. Declercq and G. Germain, Acta Crystallogr., Sect. B: Struct. Sci., 1979, 35, 2228–2229 CrossRef CAS; R. Keltjens, S. K. Vadivel, R. de Gelder, A. J. H. Klunder and T. Zwanenburg, Eur. J. Org. Chem., 2003, 1749–1758 CrossRef.
- R. Gao, W.-H. Li, P. Liu and P.-A. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o534 CAS; C. B. Nielsen, M. Pittelkow and H. O. Sorensen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o473 Search PubMed.
- S. Skovsgaard and A. D. Bond, CrystEngComm, 2009, 11, 444–453 RSC; T. Kolev, B. B. Koleva, R. W. Seidel, M. Spiteller and W. S. Sheldrick, Struct. Chem., 2009, 20, 533–536 CrossRef CAS; A. Delori, P. T. A. Galek, E. Pidcock, M. Patni and W. Jones, CrystEngComm, 2013, 15, 2916–2928 RSC.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
-
Crystal Clear-SM Expert 2.0 r11, Rigaku, 2011 Search PubMed.
-
CrysAlisPro, Agilent Technologies, Version 1.171.35.15 (release 03-08-2011 CrysAlis171.NET) Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, “X-Seed - A software tool for supramolecular crystallography”, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1435389–1435396. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02282g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
John D.
Wallis
b and
John D.
Wallis