
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterising the role of water in sildenafil citrate by NMR crystallography†
Anuji
Abraham‡
a,
David C.
Apperley
a,
Stephen J.
Byard
b,
Andrew J.
Ilott§
a,
Andrew J.
Robbins¶
a,
Vadim
Zorin||
a,
Robin K.
Harris
a and
Paul
Hodgkinson
*a
aDepartment of Chemistry, University of Durham, Durham DH1 3LE, UK. E-mail: paul.hodgkinson@durham.ac.uk
bCovance Laboratories Limited, Willowburn Avenue, Alnwick NE66 2JH, UK
First published on 21st January 2016
Abstract
A combination of solid-state NMR techniques, including 13C/1H correlation, 2H magic-angle spinning NMR and first principles calculation are employed to characterise the role of water in different hydration states of sildenafil citrate. The 13C spectrum is fully assigned for the first time and direct correlations made with respect to the crystal structure. 2H magic-angle spinning NMR is demonstrated to be a powerful tool for the study of dynamic and exchange processes in complex hydrate systems, allowing the behaviour at multiple solvate sites to be characterised without the need for expensive and selective labelling. Use of the 2H double-quantum frequency allows resolution of the different sites and, consequently, data fitting to determine rates of spin-diffusion between the different sites. The water is shown to be highly dynamic, undergoing C2 rotation, with chemical exchange between different water molecules and also with the host structure. The methods adopted are applicable to the investigation of an extensive range of hydration types found in pharmaceutical drug substances.
Introduction
Understanding the solid-state characteristics of drug substances is vital within the pharmaceutical industry. In particular, ingress of water into a drug substance or a formulated product can have significant consequences for handling, storage or product performance in vivo. Understanding the role of water in pharmaceutical hydrates is of central importance to formulation development.1 Amongst the most powerful of methods for characterising structure and dynamics in solid materials is NMR, which is particularly useful in conjunction with both computation of chemical shifts and diffraction studies, thus giving rise to the term “NMR crystallography”.2,3Sildenafil citrate (the active component of Viagra™) is a monobasic salt and, based on solution-state pKa properties, the citrate anion is expected to be deprotonated at C34. Its structure is shown in Scheme 1, together with the numbering scheme of the atoms used here. Only one polymorphic form has been reported. As discussed below, the crystal structure of this form takes up water reversibly, with no significant hysteresis or change of form on exposure to high humidity. Qualitative insights into the interaction of water with solid sildenafil citrate have previously been derived from 13C and 15N cross-polarisation magic-angle spinning (CPMAS) NMR.4 Here, variable-temperature 13C and 2H spectra, together with 13C/1H HETCOR experiments and first-principles computation, are used to develop a comprehensive picture of the interaction of water with the drug material, and to resolve discrepancies in spectral assignment with other solid-state NMR studies.5
 | ||
| Scheme 1 Molecular schematic of sildenafil citrate, indicating the numbering of carbon and nitrogen atoms used in ref. 4 and this work. Note that both molecules are shown, as conventionally represented, in their neutral forms, while XRD and NMR are consistent with a transfer of H+ from the C34 carboxylic acid group to N26 in the solid state. | ||
Experimental
Materials and methods
“Hydrated” samples of sildenafil citrate (Chemos GmbH) were prepared by storage in an enclosed container saturated with H2O or D2O for at least two weeks. Single-crystal X-ray diffraction, Dynamic Vapour Sorption (DVS) and Thermogravimetric Analysis (TGA) studies were performed on these samples. These are described in the ESI.†13C CPMAS spectra were acquired as a function of temperature on a Varian Unity Inova spectrometer operating at 299.82 MHz for 1H (75.40 MHz for 13C), with a sample of hydrated sildenafil citrate packed into a 5 mm o.d. zirconia rotor. Holes in the end-cap of the rotors avoided any risk of pressure build-up on dehydration. CPMAS spectra (at 5 kHz MAS rate) were acquired with 204 repetitions, a recycle delay of 5 s and CP contact time of 1 ms, allowing at least 20 minutes at each temperature before acquisition to allow for thermal equilibration. The order of acquisition is indicated in Fig. 1. Note that temperatures are quoted as set and do not attempt to correct for sample heating due to MAS; these corrections are estimated to range from +5 °C for 5 kHz spinning in a 5 mm rotor to a maximum of +25 K for 10 kHz spinning in a 5 mm rotor.
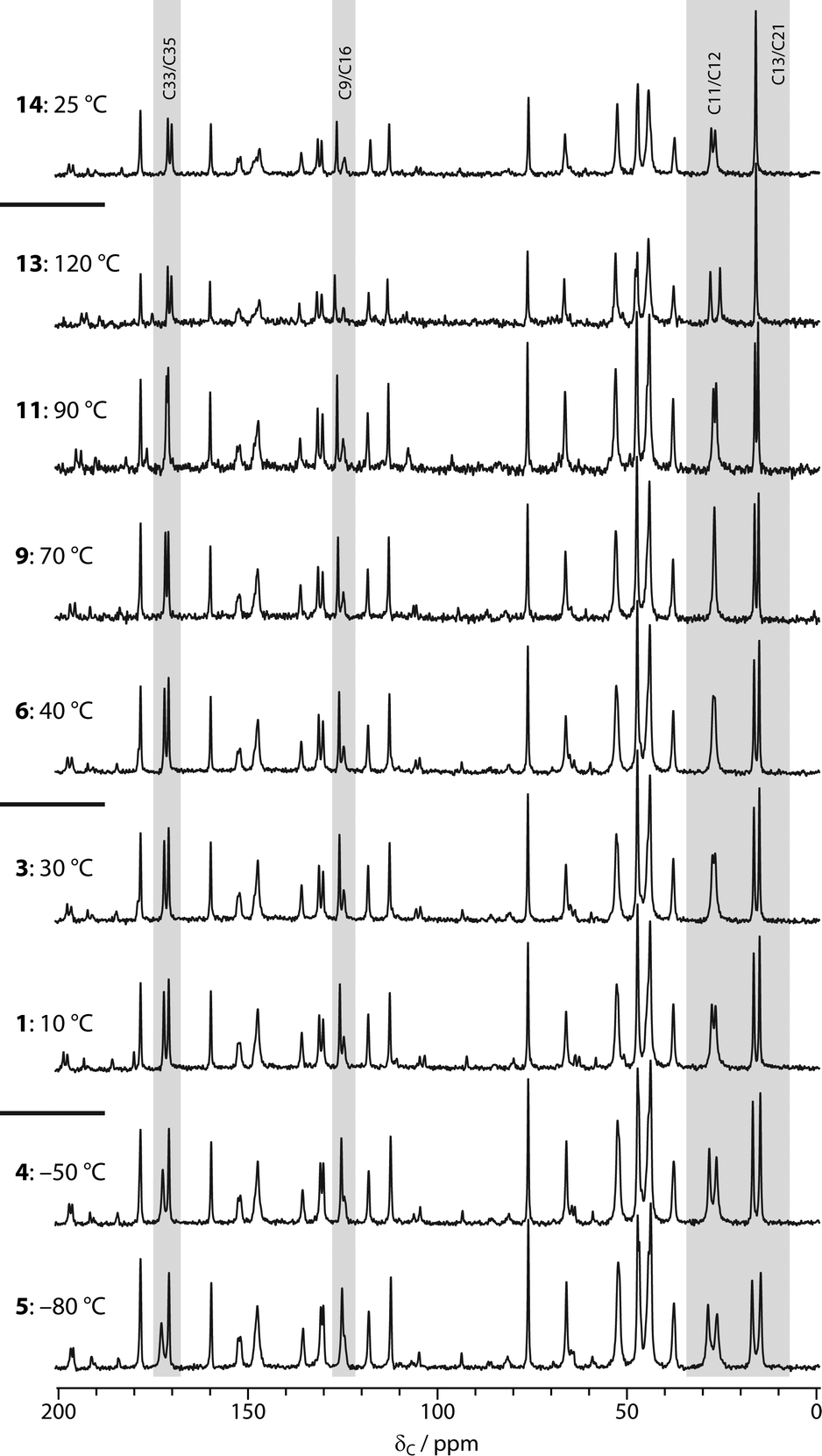 | ||
| Fig. 1 13C CPMAS spectra of initially hydrated sildenafil citrate as a function of temperature. Bold numbers indicate acquisition order, starting at 10 °C. Acquisition points 2 (20 °C), 7 (50 °C), 8 (60 °C), 10 (80 °C) and 12 (110 °C) are omitted for clarity. Shaded areas highlight spectral regions exhibiting the largest temperature dependence. | ||
13C/1H HETCOR experiments were carried out at ambient probe temperature on a Varian VNMRS spectrometer operating at 399.88 MHz for 1H (100.56 MHz for 13C), with samples packed into 4 mm o.d. zirconia rotors. The magic-angle spinning rate was 8 kHz. FSLG 1H decoupling was used in the indirect dimension at a power equivalent to 96.2 kHz, and the same power was applied for TPPM decoupling in the direct dimension. Lee-Goldburg cross-polarisation was used with both 0.1 and 1 ms contact times. 200 acquisitions were acquired with a 5 s recycle delay for each of 32 increments in t1, using a spectral width of 14.7 kHz. The 1H chemical shift axis was rescaled following a reference spectrum of glycine acquired under the same conditions (using 3.0 ppm and 8.4 ppm as the reference shifts of the CH3 and NH3+ signals respectively).
2H MAS spectra were recorded on a Varian InfinityPlus spectrometer using samples packed into 5 mm o.d. MAS rotors and at a Larmor frequency of 76.71 MHz, using a MAS rate of 8.5 kHz. 256 transients were acquired at each temperature using a recycle delay of 8 s. Most spectra were obtained via direct excitation using a 2H 90° pulse of 7.7 μs, referenced to external CDCl3 at 7.2 ppm, and using 1H TPPM decoupling. Where required, the magic angle was adjusted using either [d6]α-oxalic acid6 or [d1]-alanine to limit broadening due to magic-angle mis-set.
2H DQ/SQ EXSY spectra were acquired using the DQ/SQ correlation experiment of ref. 7, but extending the z-filter to provide the exchange period of an EXSY experiment, and using cross-polarisation for excitation. 32 transients were averaged for each of 32 t1 increments with Δt1 = 4/νrot = 400 μs, and 4 s recycle delay, using a MAS rate of 10 kHz. 1H decoupling was used by default in the acquisition dimension, although this was not observed to have a significant impact on spectral resolution. Spectra were generally acquired with “synchronised acquisition” so that spinning sidebands were folded on to centrebands in both dimensions to improve signal-to-noise ratio and quantification. A series of spectra was recorded with mixing times ranging from 1 ms to 2 s at two set temperatures, −50 and −30 °C. Note that the mixing time was actively synchronised with the rotor phase at all but the shortest mixing times.8,9 Processing of the 2D spectra involved modest Gaussian line-broadenings of 100 Hz and 50 Hz in the direct and indirect dimensions respectively prior to Fourier transformation. Further analysis was performed in Matlab,10 as described in the ESI.†
Calculations
Computations were carried out using CASTEP11 version 5.0, via Materials Studio14 version 4.4. CASTEP implements the GIPAW method12,13 using density functional theory15,16 and a plane-wave basis set to simulate the electronic wave functions, together with pseudopotentials to represent the core electrons.17 Although there is only one unit of sildenafil citrate in the asymmetric unit cell, Z = 8 (space group is Pbca), and so the full crystallographic unit cell is large. The volume of the unit cell (6425 Å3) means that a single k-point at the gamma point is sufficient to sample the Brillouin zone. A trial calculation of the NMR parameters sampling the k-space at 0.25, 0.25, 0.25 in fractional coordinates of the reciprocal lattice resulted in minor variations of the calculated 13C shieldings (generally less than 0.3 ppm) that were not significant for assignment. The exchange-correlation functional was approximated at the generalised-gradient level, specifically that of Perdew, Burke and Ernzerhof (PBE).18 Ultrasoft pseudopotentials19 consistent with the PBE approximation were generated by CASTEP on-the-fly. The planewave cut-off energies were 300 and 610 eV for geometry optimisation and NMR calculations respectively. Atomic positions were optimised using a BFGS variant20 such that the computed atomic forces were less than 0.05 eV Å−1. Lattice parameters were fixed at those given by the X-ray diffraction experiments. NMR shielding tensors were computed using the fully periodic GIPAW method.Results
X-ray diffraction, DVS and TGA
The structure derived from the single-crystal XRD study (see the ESI†) was in agreement with the previously published result21 (FEDTEO in the Cambridge Structural Database), except that the previously reported structure is presented as a simple monohydrate, without discussion of non-stoichiometry. Refining our new data with the water occupancy as a free parameter gave an occupancy of 0.494(12). To reduce the number of fitting parameters, and given that such occupancy parameters are typically strongly correlated with ADP parameters, the occupancy was fixed at 0.5 for the final refinement, corresponding to sildenafil citrate hemihydrate. Note that independent refitting of the original crystallographic data for the FEDTEO structure with a variable occupancy is reported to result in a similar occupancy factor (within one esd) and lower R factors than reported in ref. 21 (see Acknowledgements). The Dynamic Vapour Sorption (DVS) and Thermo-Gravimetric Analysis (TGA) results also support partial occupancy of the water sites. The DVS results, Fig. S1,† show a monotonic gain in sample mass of 2.16% as a function of relative humidity from 0 to 95%, which corresponds to 0.82 equivalents of H2O per sildenafil citrate at 95% RH (assuming the material is fully dehydrated at 0% RH). Previous DVS results4 gave a value of 0.78 equivalents at 90% RH, i.e. it does not appear to be feasible to achieve a true monohydrate. The mass gain of 1.78% at a typical environmental humidity of 60% would correspond to a stoichiometry of 0.67 equivalents of H2O. This is in excellent agreement with TGA measurements, which show a continuous loss of mass of 1.6% over the temperature range 40–100 °C (at a heating rate of 10 °C min−1), corresponding to loss of about 0.7 mol of H2O per mole of sildenafil citrate; no further changes occur until the onset of decomposition above 190 °C. The 2H NMR results discussed below confirm that these results are not significantly distorted by adventitious water.The DVS and TGA results show that the XRD studies are compatible, with the different reported levels of hydration reflecting differences in sample environment and differences in modelling of the disordered water. It is, therefore, a reasonable starting point for calculations to use the XRD structure with the water sites fully occupied and the same structure, but with the water atoms removed, as models for the fully hydrated and dehydrated materials respectively. The isostructural nature of the hydrated and dehydrated forms is confirmed by the calculation results below.
13C VT NMR
13C NMR has been widely used to follow the hydration behaviour of non-stoichiometric pharmaceutical solvates.22Fig. 1 shows the results of acquiring the 13C CPMAS spectra of sildenafil citrate as a function of temperature, starting with a hydrated sample at 10 °C. Changes in the spectrum are observed in three regions: 16–28 ppm, corresponding to the C11–C13 (propyl) and C21 (ethoxy) fragments of sildenafil, around 170 ppm, corresponding to the C33 and C35 resonances of citrate, and, more subtly, around 125 ppm corresponding to C9/C16 (sildenafil). Previous studies of the ambient-temperature 13C spectra of samples stored at different relative humidities had demonstrated the sensitivity of the first two of these regions to hydration state.4 This is reasonable in terms of the crystal structure, where the water molecule is hydrogen bonded to the C33 citrate and the sulphonamide group (nearest carbon C16), and is adjacent to the propyl fragment. The changes with temperature were fully reversible provided that the sample was maintained below 40 °C, as shown by the continuity of the spectral changes between 30 and 40 °C, despite the temperature having been previously cycled down to −80 °C. This implies that the ambient temperature spectrum is affected by dynamic averaging, as confirmed by the 2H NMR results below. Since the water sites are not fully occupied, and changing the water occupancy results in measurable changes to the 13C NMR spectrum, line-broadening associated with “static disorder” would be observed in the absence of exchange of water molecules between sites. In quantitative terms, the water molecules must be exchanging between occupancy sites at rates of at least 10 s of Hz at −80 °C (a 1 ppm shift difference corresponds to 75 Hz at the 13C NMR frequency used).Warming the sample to higher temperatures than 40 °C leads to further changes in the same regions of the spectrum. In contrast, however, these changes are irreversible; the final spectrum obtained at ambient temperature is identical to that of “dry” sildenafil citrate obtained by storage at 0% RH.4 The spectrum of the dehydrated material is not significantly temperature dependent, with the exception of the C11/C12 propyl resonances (compare the final spectra obtained at 120 °C and 25 °C). This suggests that the dynamics of the propyl region depend on the occupancy of the water site. Similar observations have been made via13C T1 (spin–lattice) relaxation times in the hydrated and dehydrated forms of thiamine hydrochloride monohydrate.23
2H NMR
Although less widely used than 13C NMR, 2H NMR has previously been exploited to examine water dynamics in non-stoichiometric pharmaceutical solvates, and seen to provide direct insight into local mobility. In the case of carvedilol phosphate hemihydrate, for example, a very broad pattern, was observed, consistent with the water molecules being tightly bound in the narrow channels,24 while a very narrow 2H spectrum was observed in the case of topotecan hydrochloride, consistent with very loosely constrained water molecules.25 In these cases, however, the 2H NMR spectrum signal was dominated by the water signal and static “wideline” NMR spectra were sufficient. In our case, multiple sites were observed to be deuterated, and so magic-angle spinning was used to resolve the different 2H resonances.Fig. 2 shows the 2H magic-angle spinning spectra obtained as a function of temperature. The sample was held for an extended period at 55 °C to measure 2H T1 values, which are summarised in Table S1.† It is clear from spectra acquired before and afterwards that the bulk of the dehydration occurred during this period. The low temperature spectra show three regions of intensity (in addition to any free water signal). The very broad and quickly relaxing peak at 3.7 ppm is assigned to water in sildenafil citrate tunnels. This can be readily distinguished from adventitious water, which appears as a signal around 5–6 ppm, marked with an asterisk in the dashed spectrum of Fig. 2. In contrast to the other signals, this peak is not associated with a noticeable spinning sideband manifold, and so represents a small fraction of the total water signal. This usefully illustrates that adventitious and channel water can be clearly distinguished using 2H MAS NMR, in contrast to bulk techniques such as DVS and TGA. The broadness of the peak for channel water and its extremely short relaxation time are consistent with rapid flips of the water molecules about their C2 axis. The signal around 17 ppm is readily assigned to the C33 acid H site (O33H) on the basis of the CASTEP-predicted shifts. As discussed below in the context of the two-dimensional 2H experiments, and confirmed by the HETCOR experiments, the feature around 11 ppm contains the other exchangeable H signals: the remaining citric acid protons, O35H and O31H, and the protonated nitrogen sites N5H and N26H.
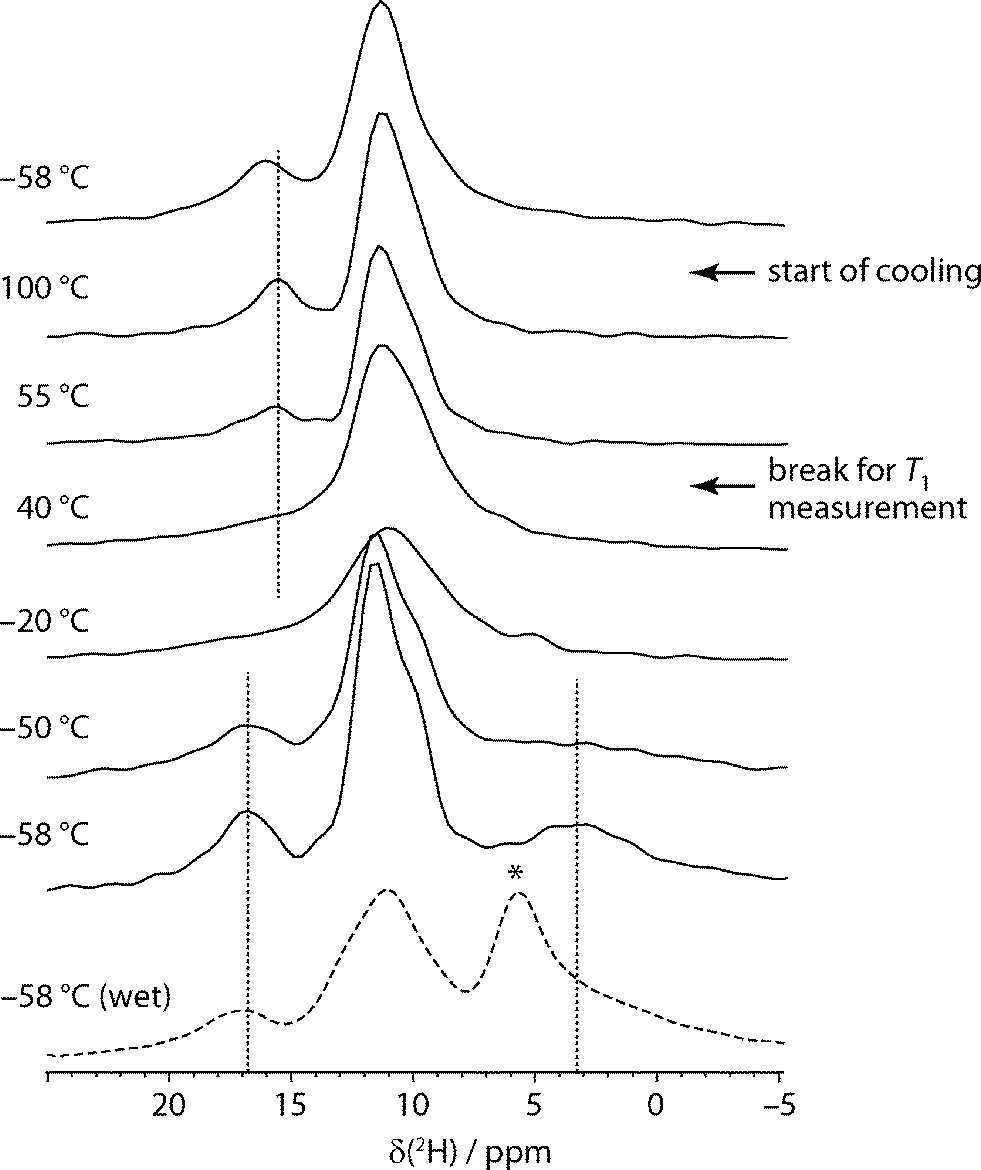 | ||
| Fig. 2 Centreband region of the direct-excitation 2H MAS spectra of deuterated sildenafil citrate acquired as a function of temperature, starting at −58 °C, heating to 100 °C before cooling to the starting temperature. Several intermediate spectra (particularly on the cooling phase) are omitted as no significant changes were observed. Dotted lines mark characteristic signals as guides to the eye. The dashed line shows the spectrum of a new “wet” sample that shows additional intensity ∼5 ppm (marked with *) assigned to adventitious water (see text). | ||
As the temperature is raised, the resolved citrate signal clearly broadens and coalesces with the water signal, which becomes too broad to be observed. Only a single signal peak at 10 ppm is seen at 10 °C, although is it difficult to judge whether a subset of, or all, the 2H sites are in exchange. The rate of exchange must be on a frequency of kHz, corresponding to collapsing a 15 ppm shift range at 76.8 MHz NMR frequency. As the temperature rises, and the sample dehydrates, the spectrum becomes less symmetric and a peak around 16 ppm appears. This corresponds to the O33H citrate signal, no longer broadened by exchange, consistent with the CASTEP calculations that show a shift of −0.5 ppm between “wet” and “dry” structures for this site. As also observed in the 13C experiments, no significant changes are observed on lowering the temperature back to the starting point, which is consistent with a lack of H-exchange/water dynamics in the dehydrated material.
As detailed in the ESI,† quadrupolar parameters were obtained for the resolved sites by fitting the 2H spectra from “wet” and dehydrated samples at −58 °C. The most informative parameters are those for the peak assigned to sequestered water. The CQ of 97 kHz and asymmetry parameter of 1 are characteristic for water molecules undergoing a rapid C2 flip, but not other significant re-orientational dynamics.26
2H DQ/SQ EXSY experiments were used to try to provide more quantitative insight into the exchange processes. Fig. 3 shows an example DQ/SQ 2H EXSY spectrum obtained at −50 °C on a “wet” sample, using cross-polarisation for excitation. As expected, using the double-quantum frequency in the indirect dimension dramatically improves resolution since this is unaffected (to first order) by the quadrupolar coupling and so is less sensitive to magic-angle mis-set and motional broadening.7 The broad peak at 10–12 ppm is clearly resolved into a smaller peak about 11.5 ppm and a large peak about 10 ppm. The HETCOR-derived and computed hydrogen shifts of N5H, N26H, O31H and O35H are too overlapped to confidently assign the peaks. The peak at 11.5 ppm is tentatively ascribed to the other citrate carboxylic acid proton, O35H, on the basis of its similar intensity to the O33H peak and its calculated proton chemical shift. The use of cross-polarisation allowed short recycle delays, but resulted in spectra without an observable signal from D2O. Use of direct-excitation, rather than cross-polarisation, to observe EXSY spectra containing a water signal was unsuccessful, however; it was difficult to excite a water double-quantum signal in the DQ/SQ experiment, and at short mixing times (e.g. 7.1 ms) strongly distorted 2D spectra were obtained in the simple SQ/SQ version of the experiment. This presumably relates to the rapid C2 flips of the water, violating the EXSY requirement that the NMR frequency is stable during the t1 and t2 measurement periods. At long mixing times (e.g. 71.4 ms), undistorted spectra were observed, but without a water resonance, as the troublesome water signal has been lost through relaxation (a T1 of 37 ms was measured at −35 °C).
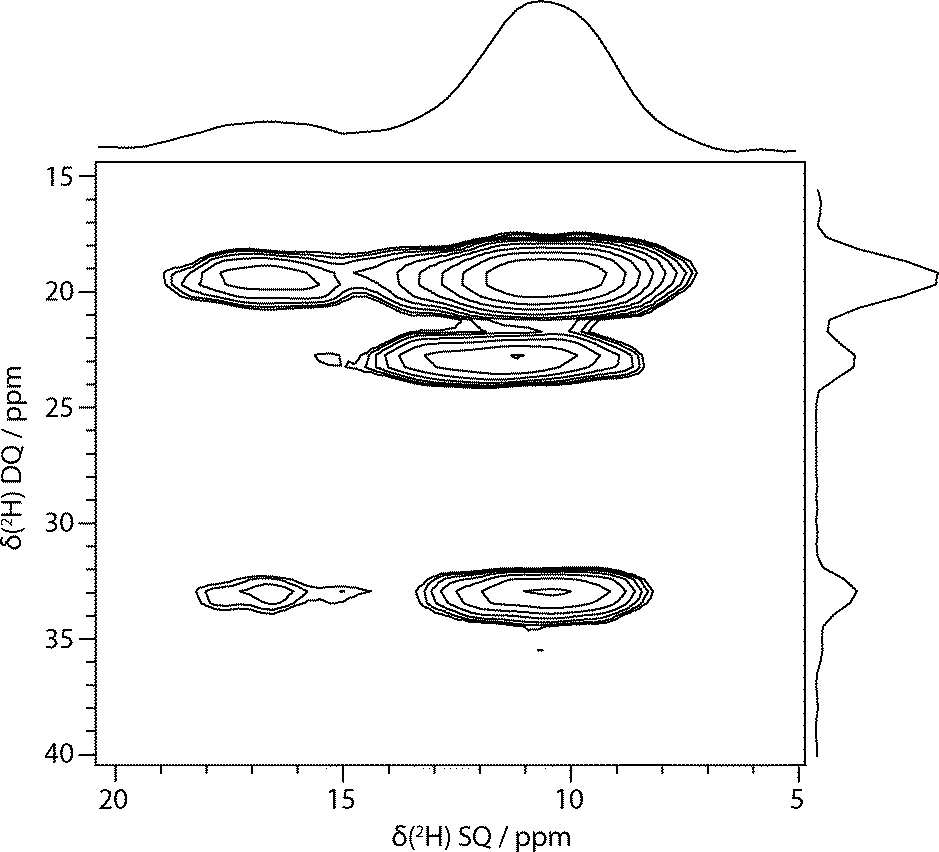 | ||
| Fig. 3 Example 2H DQ-SQ EXSY spectrum of sildenafil citrate hydrated with D2O acquired at −50 °C, 1 s mixing period, using cross-polarisation from 1H for excitation and synchronised acquisition in both dimensions. | ||
Exploiting the additional site resolution provided by the DQ/SQ experiment, the build-up of the EXSY cross-peaks was used to estimate rates of magnetisation exchange between the sites, following the methodology described in the ESI.† As illustrated in Fig. 4, very satisfactory fits were obtained. The rates of exchange were found, however, to be identical within experimental uncertainty for the two measurement temperatures, as summarised in Table S4.† This implies that magnetisation transfer in these experiments was dominated by 2H spin-diffusion rather than chemical exchange. It has previously been observed that magic-angle spinning tends to enhance rates of magnetisation exchange via dipolar couplings between 2H sites.40 As noted in the discussion of the data in Table S4,† measurement of 2H spin-diffusion rates might provide useful information on spatial proximity, in the same way that 1H spin-diffusion has been used to provide constraints for structure solution.27,28 In this case, however, the overall structure is known and so the spin-diffusion results provide little additional insight.
 | ||
| Fig. 4 Intensities of cross-peaks in 2H DQ/SQ EXSY spectrum and corresponding fits obtained on hydrated sildenafil citrate at −50 °C. Columns correspond to frequencies in the direct (single-quantum) dimension, and rows to indirect dimension (double-quantum) frequencies. Error bars indicate one-standard deviation uncertainties on the intensity values. The corresponding figure for data acquired at −30 °C can be found as Fig. S3 of the ESI.† | ||
First principles calculations
The two starting points for calculation were a “wet” structure, based on the initial XRD-derived structure with fully occupied water sites, and a “dry” structure with the water atoms removed. A number of different geometry optimisations were performed prior to calculation of the NMR parameters using lattice parameters fixed at their XRD-derived values: optimisation of H positions only (“wet” and “dry” structures), optimisation of all atomic positions (“wet” structure only) and optimisation of just the citrate ion atoms (“dry” structure only). All-atom optimisation was not attempted on the artificial and potentially unstable structure obtained by removing the water molecules.Experimental 13C shifts were obtained from the spectra of dehydrated and hydrated samples at ambient probe temperature and −80 °C respectively (results tabulated in the ESI†). In cases where resonances overlapped (e.g. C25 and C27), the same experimental shift was assumed for the different sites. The detailed assignment of the experimental chemical shifts is discussed below in the context of the HETCOR experiments.
In all cases, excellent correlations are obtained between calculated and experimental 13C chemical shifts, cf. Fig. S4.† As typically observed, the slope of the fitted linear gradient is not exactly 1. Such deviations in slope may be due to the effects of nuclear delocalisation29 and/or systematic errors in the DFT methodology30 and cannot be readily calculated. We have taken the usual pragmatic approach of rescaling the calculated shifts to match the slope and mean of the experimental shifts.31 This has been done independently for each computed result prior to calculating the root-mean-square deviation between experimental and calculated shifts. As summarised in Table 1, there is little difference in the overall RMSD between experimental and calculated shifts for the different geometry-optimised shifts. Note that the citrate shifts are not particularly well-fitted by H-only optimisation, and the RMSD for these signals is significantly reduced when the position of the citrate atoms is allowed to “relax”. The anisotropic displacement parameters for several of the non-hydrogen citrate atoms are significantly elongated, suggesting the presence of (dynamic) disorder which may slightly distort the average citrate geometry in the starting crystal structure. Similar behaviour was observed for disordered acetic acid molecules in the diacetic acid solvate of diterbutaline sulfate.32
| Geometry optimisation | “Wet” | “Dry” | ||
|---|---|---|---|---|
| All | Citrate | All | Citrate | |
| H-only | 1.6 | 2.9 | 1.6 | 2.8 |
| H + citrate | 2.0 | 0.8 | ||
| All atoms | 1.8 | 1.4 | ||
Fig. 5 compares the differences in 13C shift between the hydrated and dehydrated sildenafil citrate resonances observed both experimentally and in computation. Experimental shifts were obtained from dehydrated and hydrated samples at ambient probe temperature and −80 °C respectively, while calculated shift differences are based on the (rescaled) shifts of the two H-only optimised structures. The sign of the changes is generally well predicted, although the calculations tend to overestimate their magnitude, which is likely to reflect the partial occupancy of the water sites in the hydrated sample. The possible effects of fractional water occupancy on 13C chemical shifts have previously been noted for the hemihydrate of a guanosine derivative.33 The agreement for sites that show significant temperature dependence and are likely to be affected by dynamics, e.g. C11 and C21, is noticeably poorer. This is expected given that the calculations are based on a single 0 K optimised structure. The agreement is far better than would be expected if the “errors” between calculated and experimental shifts were uncorrelated; the dotted lines in Fig. 5 are placed at  ×1.6 ppm, where 1.6 ppm corresponds to the typical RMSD observed in Table 1. These results indicate that first-principles calculations can be used to understand relatively subtle differences for sites in otherwise equivalent environments. Significantly poorer correlations between wet vs. dry chemical shifts are obtained if either of the more fully optimised structures was used, i.e. the effect of the presence of water on the chemical shifts is better predicted when the non-H atoms are fixed in consistent positions.
×1.6 ppm, where 1.6 ppm corresponds to the typical RMSD observed in Table 1. These results indicate that first-principles calculations can be used to understand relatively subtle differences for sites in otherwise equivalent environments. Significantly poorer correlations between wet vs. dry chemical shifts are obtained if either of the more fully optimised structures was used, i.e. the effect of the presence of water on the chemical shifts is better predicted when the non-H atoms are fixed in consistent positions.
 | ||
| Fig. 5 Calculated (blue) vs. experimentally observed (red) differences in 13C chemical shift between dehydrated and hydrated sildenafil citrate. The shifts of overlapped peaks, where the assignment is ambiguous, have been averaged as indicated. Sites affected by dynamics are marked by paler colours and italic labels. Dotted lines indicated expected one-standard deviation variance expected if individual “errors” are uncorrelated. | ||
13C/1H HETCOR
Fig. 6 shows the superimposed HETCOR spectra obtained at long mixing times for the wet and dry samples. Extracted 1H chemical shifts are tabulated in Table S5.† These were determined from the cross-peaks at short-contact time, where present, or otherwise from clear and distinctive long-range correlation peaks, noting that small differences can be observed in apparent 1H shifts from short vs. long-range correlation peaks.34,35 Deuterated samples prepared by recrystallisation from D2O or exchange in a D2O-saturated atmosphere had identical 13C NMR spectra. HETCOR spectra of either deuterated sample showed no correlation peaks above 1H shifts of 10 ppm, indicating that all exchangeable hydrogens on the citrate and N5/N26 of the sildenafil are indeed exchanged.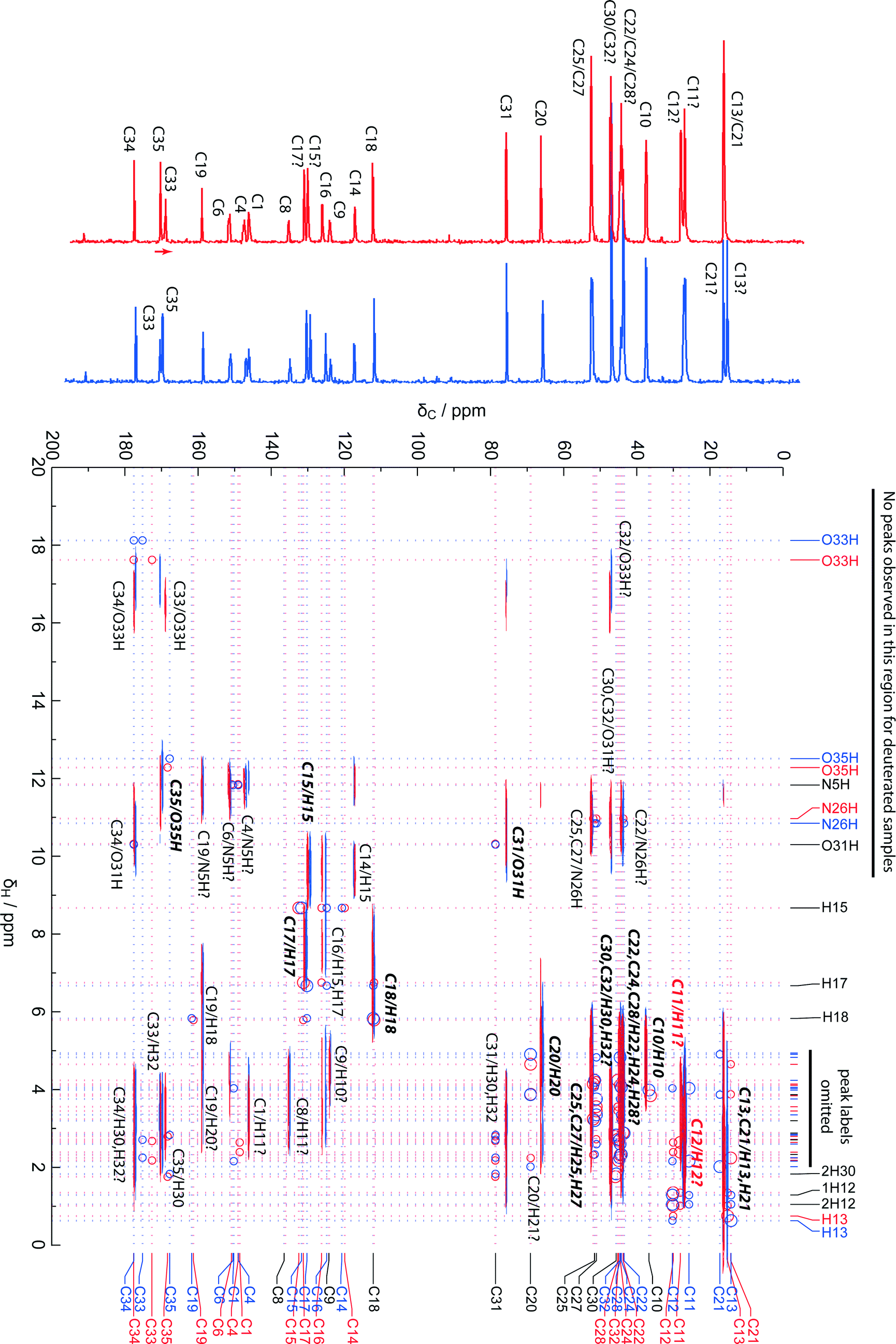 | ||
| Fig. 6 Superimposed 13C/1H HETCOR spectra of hydrated (blue) and dehydrated (red) samples at 1 ms mixing time. One-dimensional 13C spectra are skyline projections. Red and blue dotted lines mark shifts derived from calculated isotropic shielding values (H-only optimisation) using approximate references to align experimental and calculated shifts (170 ppm for 13C, 31 ppm for 1H). Separate red and blue labels are combined to a single black label where computed shifts are very similar. Circles show predicted correlation peaks, with the circle size reflecting the C, H distance: large circles less than 1.5 Å, small circles between 1.5 and 2.5 Å. Peaks that also appear in the spectrum with 100 μs mixing time are marked in bold italics. | ||
Also shown in Fig. 6 are the corresponding computed chemical shifts based on the H-only geometry-optimised structures, with circles marking cross-peaks expected on the basis of distances between each carbon site and the nearest hydrogens, obtained using the crystal-viewing software gdis.36 Larger circles mark distances below 1.5 Å, corresponding to directly bonded CH pairs. In all cases, corresponding cross-peaks (marked in bold italics) were observed in the short-contact-time HETCOR experiment. A few longer-range contacts, corresponding to distances less than 2.0 Å, were also observed at these short mixing times. These cross-peaks were helpful in a number of cases. For example, the pair of lines at 130 and 131 ppm can be assigned to C15 and C17 using the predicted chemical shifts of the attached H sites, which differ by ∼2 ppm. This assignment disagrees with the ordering predicted from the computed 13C shifts alone, but is within the approximately 1.6 ppm “margin of uncertainty” suggested by the overall RMSD between computed and observed 13C shifts. In other areas, such as C30 and C32, the 13C and 1H shifts are both too strongly overlapped to allow the assignment to be improved.
The additional cross-peaks appearing at longer mixing time help to resolve most of the remaining ambiguities. Smaller circles in Fig. 6 mark potential cross-peaks corresponding to distances up to 2.5 Å. For example, long-range correlations to O33H and O35H hydrogen sites allow C33 vs. C35 to be assigned. The assignments are confirmed by the changes in the shifts of these peaks with hydration state. A small number of cross-peaks seem to correspond to even longer distances (up to 2.8 Å), but this may be associated with spin-diffusion of magnetisation between H sites and should be interpreted cautiously. For example, the apparent cross-peak between C31 and O33H is more likely to be a “relay” transfer than a direct correlation. Taken together, however, the computed 13C shifts, short-contact cross-peaks identifying H peaks, longer range correlations, and the peak movements between wet and dry samples allow all the resolved 13C peaks to be assigned with some confidence. The “evidence” used to assign each 13C peak is summarised in Table S5.† Overall, the availability of powerful computational tools, together with experimental HETCOR results, has allowed the sildenafil citrate assignment to be considerably refined compared to earlier work.4,5 First-principles calculations and HETCOR experiments have recently been used in a similar fashion to resolve conflicting spectral assignments for the API naproxen,37 and aid structure solution from PXRD data for an indomethacin/nicotinamide co-crystal.38 Note that Wawer et al.5 have different assignments for the solution-state NMR resonances in the 135–155 ppm region compared to ref. 4, inverting the assignments for the pairs C4/C6 and C1/C8 in the numbering used here. The current solid-state study also reverses the order of C4/C6 compared to our previous work, but not for C1/C8. The difference in shift between C4 and C6 (<4 ppm) is sufficiently small, however, to be ascribed to differences in shift between solid and solution states.
Conclusions
The use of first-principles calculation to connect diffraction-derived crystal structures with solid-state NMR observables has allowed a full assignment of the resolved 13C resonances of a complex pharmaceutical solid. The differences in chemical shift upon hydration are relatively small; for comparison, changes up of to 2 ppm and 5 ppm were observed in 1H and 13C chemical shifts in a study of ciprofloxacin.39 Although they are smaller than the overall RMSD between calculated and experimental 13C shifts, a clear correlation was observed between the calculated and experimental shifts on hydration, confirming their significance and utility in assignment. The fuller assignment allows the effects of hydration on the 13C chemical shifts of the citrate ion to be quantified, where the assignment was previously unknown. Computed 1H shifts were also significant in assignment, particularly in the less crowded regions above 6 ppm. Subtle differences in the propyl region of the 13C spectrum could be explained by differences in dynamics depending on occupancy of the water sites.The most direct insight into hydration was provided by 2H MAS NMR, which allows multiple exchangeable sites to be studied simultaneously, without the need for bespoke isotopic labelling. The water molecules are found to be undergoing C2 flips and exchanging between water sites (information obtained from the 13C spectrum), even at the lowest temperatures studied (−58 °C). This is in good agreement with the XRD crystal structure, in which the water hydrogen atoms were located, but in which water sites were partially occupied. In addition, exchange of hydrogen between water and other exchangeable hydrogen sites, most evidently on the citrate ion, was observed as the temperature was raised. This is consistent with the easy deuteration of exchangeable sites from storing sildenafil citrate in a D2O-saturated atmosphere. The sensitivity of the 2H NMR signal to dynamics complicated, however, the use of magnetisation transfer experiments to measure the rate of this hydrogen exchange. It was, nevertheless, possible to derive rates of 2H spin-diffusion from double-quantum/single-quantum EXSY experiments, which may be useful in cases where the 2H MAS spectrum is sufficiently resolved.
Aided by computation, solid-state NMR has provided a detailed picture of the interaction of water molecules with the host sildenafil citrate structure in this non-stoichiometric hydrate. Although molecular dynamics simulations have allowed relatively slow molecular processes, on the order of microseconds, to be linked to NMR observations,41 reproducing the behaviour observed here in simulation will be extremely challenging, due to the size of the system (696 atoms in the unit cell) and the electronic re-arrangement involved in hydrogen exchange.
Acknowledgements
The authors thank Dr. Jonathan Yates for initial assistance with the CASTEP calculations and Dr. Dmitry S. Yufit from the Durham Chemistry crystallography service. Crystallographic data for the sildenafil citrate structure have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC-1062242. We are grateful to one referee for forwarding the results of a refitting of the crystallographic data from ref. 21.This work was supported under EPRSC grant number EP/D057159, and benefitted from discussions within the context of the EPSRC-funded collaborative computational project for NMR crystallography (CCPNC). Raw data and supporting electronic files associated with this publication can be accessed through data DOI: 10.15128/rf55z7713, as summarised in Table S6 of the ESI.†
Notes and references
- K. R. Morris, in “Polymorphism in Pharmaceutical Solids”, ed. H. G. Brittain, Marcel Dekker, New York, 1999, pp. 125–181 Search PubMed.
- “NMR Crystallography”, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, Wiley, 2009 Search PubMed.
- L. Mafra, Solid State Nucl. Magn. Reson., 2015, 65, 1 CrossRef PubMed.
- D. C. Apperley, P. A. Basford, C. I. Dallman, R. K. Harris, M. Kinns, P. V. Marshall and A. G. Swanson, J. Pharm. Sci., 2005, 94, 516 CrossRef CAS PubMed.
- I. Wawer, M. Pisklak and Z. Chilmonczyk, J. Pharm. Biomed. Anal., 2005, 38, 865 CrossRef CAS PubMed.
- S. Antonijevic and G. Bodenhausen, Angew. Chem., Int. Ed., 2005, 44, 2935 CrossRef CAS PubMed.
- M. Cutajar, S. E. Ashbrook and S. Wimperis, Chem. Phys. Lett., 2006, 423, 276 CrossRef CAS.
- A. F. de Jong, A. P. M. Kentgens and W. S. Veeman, Chem. Phys. Lett., 1984, 109, 337 CrossRef CAS.
- M. R. Hampson, J. S. O. Evans and P. Hodgkinson, J. Am. Chem. Soc., 2005, 127, 15175 CrossRef CAS PubMed.
- MATLAB version 7.7.0, The MathWorks Inc., Natick, Massachusetts, 2008 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- Materials Studio version 4.4, Accelrys, Inc., San Diego, 2010 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- B. G. Pfrommer, M. Cote, S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233 CrossRef CAS.
- H. S. Yathirajan, B. Nagaraj, P. Nagaraja and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o489 CAS.
- R. K. Harris, Analyst (Cambridge, U. K.), 2006, 131, 351 RSC.
- R. L. Te, U. J. Griesser, K. R. Morris, S. R. Byrn and J. G. Stowell, Cryst. Growth Des., 2003, 3, 997 CAS.
- F. G. Vogt, R. C. B. Copley, R. L. Mueller, G. P. Spoors, T. N. Cacchio, R. A. Carlton, L. M. Katrincic, J. M. Kennady, S. Parsons and O. V. Chetina, Cryst. Growth Des., 2010, 10, 2713 CAS.
- F. G. Vogt, J. Brum, L. M. Katrincic, A. Flach, J. M. Socha, R. M. Goodman and R. C. Haltiwanger, Cryst. Growth Des., 2006, 6, 2333 CAS.
- K. Larsson, J. Tegenfeldt and K. Hermansson, J. Chem. Soc., Faraday Trans., 1991, 87, 1193 RSC.
- B. Elena and L. Emsley, J. Am. Chem. Soc., 2006, 127, 9140 CrossRef PubMed.
- C. J. Pickard, E. Salager, G. Pintacuda, B. Elena and L. Emsley, J. Am. Chem. Soc., 2007, 129, 8932 CrossRef CAS PubMed.
- M. Dračínský and P. Hodgkinson, Chem. – Eur. J., 2014, 20, 2201 CrossRef PubMed.
- R. Laskowski, P. Blaha and F. Tran, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195130 CrossRef.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, V. Zorin and J. R. Yates, Magn. Reson. Chem., 2007, 45, S174 CrossRef CAS PubMed.
- D. C. Apperley, A. S. Batsanov, S. J. Clark, R. K. Harris, P. Hodgkinson and D. B. Jochym, J. Mol. Struct., 2012, 1015, 192 CrossRef CAS.
- G. N. M. Reddy, D. S. Cook, D. Iuga, R. I. Walton, A. Marsh and S. P. Brown, Solid State Nucl. Magn. Reson., 2015, 65, 41 CrossRef CAS PubMed.
- R. K. Harris, P. Hodgkinson, V. Zorin, J.-N. Dumez, B. Elena, L. Emsley, E. Salager and R. Stein, Magn. Reson. Chem., 2010, 48, S103 CrossRef CAS PubMed.
- A. S. Tatton, I. Frantsuzov, S. P. Brown and P. Hodgkinson, J. Chem. Phys., 2012, 136, 084503 CrossRef PubMed.
- S. Fleming, gdis, http://gdis.sourceforge.net Search PubMed, Last accessed: 2015-04-28.
- J. Czernek, Chem. Phys. Lett., 2015, 619, 230 CrossRef CAS.
- D. V. Dudenko, P. A. Williams, C. E. Hughes, O. N. Antzutkin, S. P. Velaga, S. P. Brown and K. D. M. Harris, J. Phys. Chem. C, 2013, 117, 12258 CAS.
- L. Mafra, S. M. Santos, R. Siegel, I. Alves, F. A. Almeida Paz, D. Dudenko and H. W. Spiess, J. Am. Chem. Soc., 2012, 134, 71 CrossRef CAS PubMed.
- M. Cutajar, M. H. Lewis and S. Wimperis, Chem. Phys. Lett., 2007, 449, 86 CrossRef CAS.
- A. J. Ilott, S. Palucha, A. S. Batsanov, M. R. Wilson and P. Hodgkinson, J. Am. Chem. Soc., 2010, 192, 5179 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of XRD, TGA and DVS studies; details of 2H NMR studies; detailed information on peak assignment; contents of raw data bundle. CCDC 1062242. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02234g. |
| ‡ Current address: Drug Product Science & Technology, Bristol-Myers Squibb, 1 Squibb Drive, New Brunswick, New Jersey 08903, USA. |
| § Current address: Department of Chemistry, New York University, 100 Washington Square East, New York, New York 10003, USA. |
| ¶ Current address: Solid-Form Solutions, 1 The Fleming Building, Edinburgh Technopole, EH26 0BE, UK. |
| || Current address: ExxonMobil Chemical Europe Inc., Hermeslaan 2, 1831 Machelen, Belgium. |
| This journal is © The Royal Society of Chemistry 2016 |