
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-induced self-assembly of positively charged polycyclic aromatic hydrocarbons towards nanostructures with controllable two-dimensional morphologies†
Chongqing
Yang
a,
Dongqing
Wu
*a,
Wuxue
Zhao
a,
Weizhen
Ye
b,
Zhixiao
Xu
a,
Fan
Zhang
a and
Xinliang
Feng
ac
aSchool of Chemistry and Chemical Engineering, Shanghai JiaoTong University, 200240 Shanghai, China. E-mail: wudongqing@sjtu.edu.cn
bResearch Institute of Petroleum Processing, SINOPEC, 100083 Beijing, China
cDepartment of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 4th January 2016
Abstract
A controllable self-assembly strategy of positively charged polycyclic aromatic hydrocarbons (PCPAH) towards the formation of rectangle sheets and ribbon-like nanostructures has been achieved by choosing divalent anions with different sizes. In contrast, only rod-like nanostructures are obtained from PCPAH with univalent anions. It is revealed that the divalent anions play a key role in guiding the packing of PCPAH, which provides an unprecedented route to fabricate two-dimensional nanostructures.
Over the last decade, the enthusiasm of scientific researchers towards two-dimensional (2D) nanomaterials has been boosted by graphene and its analogues due to their unique flat morphology and anisotropic chemical/physical behavior.1 However, the delicate fabrication of 2D materials with desired compositions and properties is still challenging.2 In this respect, the self-assembly of organic molecules provides a mild and solution processable strategy for the bottom-up fabrication of free-standing 2D soft nanomaterials.3 More importantly, the chemical and physical properties of these organic nanosheets,4 including electronic/photonic properties,5 porosity6 and metal or protein recognition activities7 can be precisely defined by the rational design of molecular building blocks.
Combining the extended aromatic frameworks with the incorporation of a charged heteroatom, PCPAH offer both strong π–π interactions to enhance the molecular stacking and good affinity with polar solvents, making them attractive building blocks in supramolecular chemistry.8 Thus, one can tune the self-assembly behavior and optoelectronic properties of PCPAH via the modification of their aromatic core, substituents and associated anions, which render them highly desirable candidates for the bottom-up synthesis of 2D soft nanomaterials. In our previous work, it was found that a typical PCPAH molecule, the 2-phenyl-9-benzo[8,9]quinolizino[4,5,6,7-fed]phenanthridinylium salt (PQP) containing a tetradecyl chain (shortened as PQPC14), can spontaneously self-organize into 2D ribbon-like nanostructures in solution with the short and rigid disulfonate anion serving as the cross-linker to direct the orientation of the neighbouring PQPC14 cations. More importantly, the ribbon morphologies of the PQPs exhibited strong dependence on the rigidity and valences of the organic anions.9 Inspired by these results, we envision that the proper selection of inorganic divalent counteranions may also guide the packing behavior of PQPs with short alkyl chains and facilitate the formation of nanostructures with controlled 2D sheet-like morphologies.
Herein, the self-assembly behavior of hexyl-substituted PQPs (PQPC6, Scheme 1) with divalent anions including tetrachloroplatinate (PtCl42−) and hexachloropalladate (PdCl62−) as well as univalent tetrachloroaurate (AuCl4−) in a binary solvent system (DMF/MeOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) was investigated. Interestingly, rectangle nanosheets were formed from PQPC6 salts employing PtCl42− as the counteranion. Moreover, the control over the packing of PQPC6 cations could be further achieved by tuning the steric hindrance and valence of the counteranions with PdCl62− and AuCl4−, resulting in morphology variation from 2D ribbons to one-dimensional (1D) rods. Based on these results, it is revealed that the divalent anions (PtCl42− and PdCl62−) played a key role in adjusting the non-covalent interactions between the adjacent PQPC6 cations, thus giving rise to the unequal growth rates of the aggregates along different dimensions.
:7) was investigated. Interestingly, rectangle nanosheets were formed from PQPC6 salts employing PtCl42− as the counteranion. Moreover, the control over the packing of PQPC6 cations could be further achieved by tuning the steric hindrance and valence of the counteranions with PdCl62− and AuCl4−, resulting in morphology variation from 2D ribbons to one-dimensional (1D) rods. Based on these results, it is revealed that the divalent anions (PtCl42− and PdCl62−) played a key role in adjusting the non-covalent interactions between the adjacent PQPC6 cations, thus giving rise to the unequal growth rates of the aggregates along different dimensions.
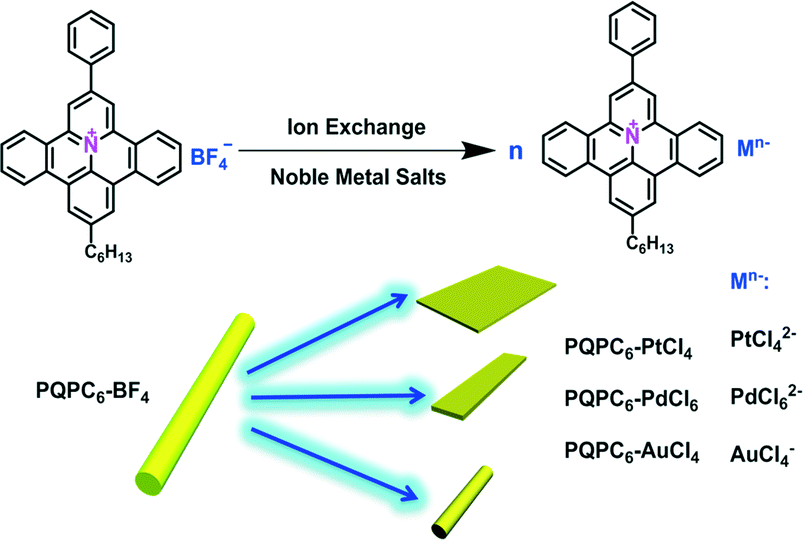 | ||
| Scheme 1 The ion-exchange route for preparing PQPC6-based ionic complexes with different counter ions. | ||
The ionic complexes of PQPC6–M (PQPC6–PtCl4, PQPC6–PdCl6 and PQPC6–AuCl4, Scheme 1) were prepared from PQPC6–BF4 with potassium tetrachloroplatinate (K2PtCl4), potassium hexachloropalladate (K2PdCl6) and potassium tetrachloroaurate (KAuCl4) in a one-to-one charge ratio via a facile ion-exchange procedure.10 Typically, PQPC6–BF4 was first dissolved in a binary solvent system (DMF/MeOH 1:7) and the aqueous solution containing the potassium salts of the corresponding anions was then added drop-wise (see the ESI†). Due to the amphiphilic nature of the ionic complex, aggregates with an ordered packing in such a mixed solvent were obtained as direct precipitate from the solution. The compositions of the obtained three ionic complex aggregates were confirmed by the elemental analysis results (Table S1†) and Fourier transform infrared (FTIR) spectra (Fig. S1†).
The morphology of the aggregates from PQPC6–PtCl4 was first investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1a and b, rectangle-shaped 2D nanosheets with sharp edges were obtained. The width of the nanosheets is between 200 and 400 nm, while the length is in the range of 200 nm–1 μm. The atomic force microscopy (AFM) image (Fig. 1c) reveals that the nanosheets of PQPC6–PtCl4 have a smooth surface with a height of ∼23 nm, further confirming their 2D morphology. It should be noted that the mixed solvent in this work is essential for the formation of the above mentioned 2D aggregates and that the aggregates do not form when either DMF or MeOH alone was used as the solvent (Fig. S2†).
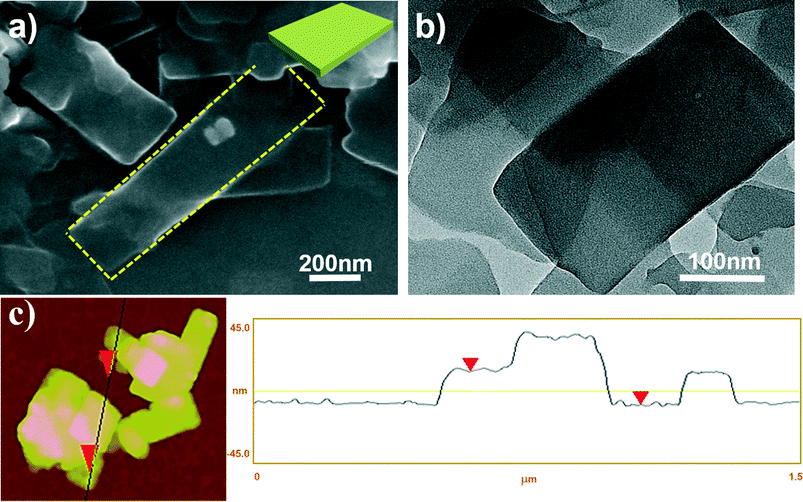 | ||
| Fig. 1 (a) SEM and (b) TEM images; (c) AFM image and height profile of rectangular nanosheets formed by ionic complex PQPC6–PtCl4. | ||
The wide-angle X-ray diffraction (WAXD) patterns of PQPC6–PtCl4 show intense peaks at 4.2, 7.6, 8.5 and 9.7° (Fig. 2a), corresponding to the (100), (101), (200) and (010) facets of a monoclinic packing, respectively. The diffraction peak at approximately 25.2° is attributed to the π–π stacking of the aromatic frameworks of PQPC6 cations.9 Based on these results, Pawley refinement on the Reflux Plus module of the Materials Studio program version 6.1 was subsequently executed to calculate the unit cell parameters of PQPC6–PtCl4 in the rectangular nanosheets.11 With the profile-fitting factors wRp and Rp being 6.35 and 4.58 % respectively, the unit cell parameters of PQPC6–PtCl4 are calculated to be a = 23.10 Å, b = 9.08 Å, and c = 11.70 Å (Table S2†).
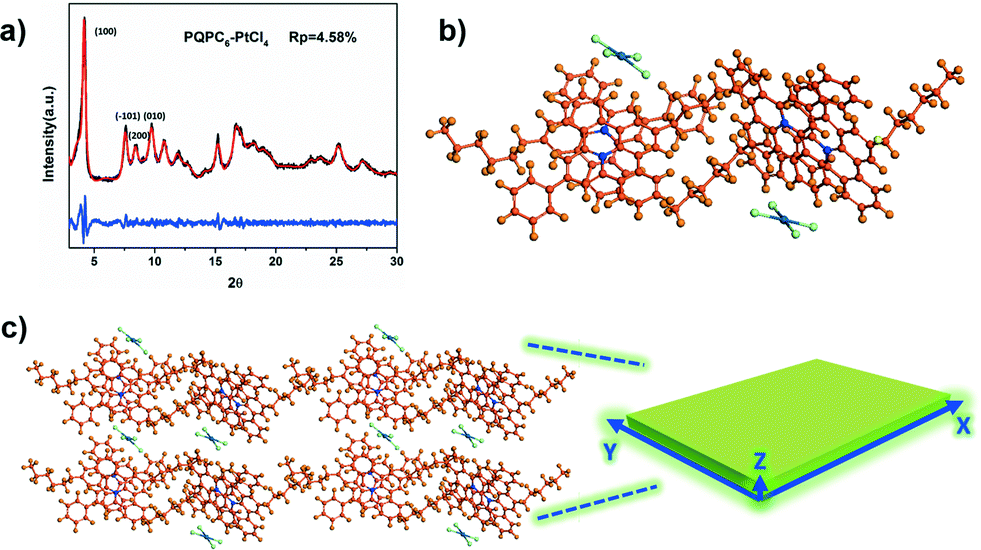 | ||
| Fig. 2 (a) The experimental WAXD pattern (black), the Pawley refined one (red) and a difference plot (blue) of PQPC6–PtCl4; (b) the possible packing model of PQPC6–PtCl4 with a slipped head-to-head arrangement; (c) the theoretical 2D staggered structure of the PQPC6–PtCl4 aggregates (orange: C; blue: N; green: Cl; dark green: Pt) and the schematic illustration of the 2D nanostructures from PQPC6–PtCl4. | ||
Accordingly, the packing behavior of PQPC6–PtCl4 in the rectangular nanosheets could be proposed by considering the previously reported packing mode of PQPs with different counteranions.8b,9 As illustrated in Fig. 2b, two neighbouring PQPC6 cations can form a dimer structure in the aggregates with their aromatic core arranged in a slipped head-to-head manner and the PtCl42− anion shared by two cations located at their bay position. Driven by the local micro-phase separation between their soft and rigid parts, these dimers can further stack together via non-covalent forces including π–π interactions, ionic interactions, etc.8c
The simulated packing model from a quantum chemical calculation on CASTEP (Cambridge Sequential Total Energy Package) suggests that the stacking of the molecules along the three dimensions is mainly dominated by three kinds of non-covalent forces: solvophobic interactions along the X-axis, electrostatic effects between the PQPC6+–PtCl42− ion pairs along the Y-axis, and the π–π interactions along the Z-axis (Fig. 2c). Under polar experimental conditions, the solvophobic interactions between the alkyl moieties will be enhanced along the X-axis, and the strong electrostatic interactions between the cation–anion pairs greatly inhibit the repulsion between the adjacent dimers along the Y-axis.12 However, the centrally charged aromatic core would prefer to expose themselves to the solvent, thus causing a slipped face-to-face structure between the adjacent cations and greatly reducing π–π interactions along the Z-axis. Driven by these interactions, the packing of the PCPAH in the direction parallel to the X–Y interfaces should be much faster than that in the direction along the Z-axes, hence leading to the formation of 2D nanostructures.
The formation of 2D nanostructures from PQPC6–PtCl4 inspired us to further study the self-assembly behavior of PQPC6–PdCl6, which also contains a divalent complex anion with a larger volume (ionic radius PtCl42− = 3.07 Å, PdCl62− = 3.33 Å).13 As demonstrated in the SEM and TEM images, the aggregates of PQPC6–PdCl6 are uniform nanoribbons with a length of 600 nm to several micrometres and a width of 100 to 300 nm (Fig. 3a and b). AFM characterization shows that the thickness of the nanoribbons is around 47 nm (Fig. 3c).
 | ||
| Fig. 3 (a) SEM and (b) TEM images; (c) AFM image with a height profile from the 2D nanoribbons formed by PQPC6–PdCl6. | ||
Different from PQPC6–PtCl4, the WAXD patterns of PQPC6–PdCl6 exhibit obvious diffractions at 6.06, 9.00, 10.80 and 12.12°, which are typical for the (001), (010), (101) and (002) facets of a triclinic packing. Additionally, the diffraction at 25.4° is attributed to the π–π interactions among the aromatic frameworks of PQPC6 cations. The lattice parameters of the triclinic space group formed by PQPC6–PdCl6 are calculated as a = 8.40 Å, b = 14.93 Å and c = 21.39 Å with the wRp and Rp converged to 12.80 and 9.26 %, separately (Fig. 4a).
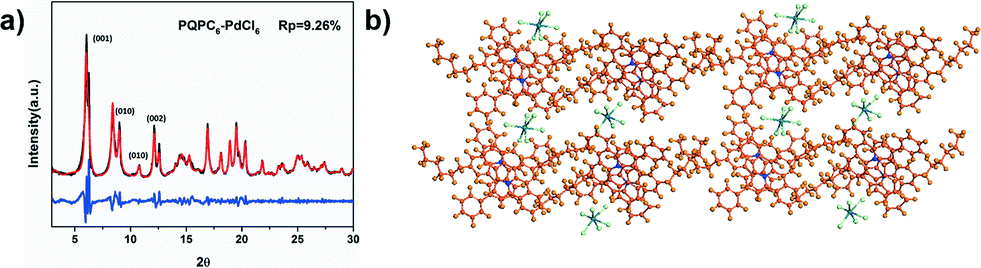 | ||
| Fig. 4 (a) The experimental WAXS pattern (black), the Pawley refined one (red) and a difference plot (blue) of PQPC6–PdCl6; (b) the theoretical possible 2D staggered structure of the PQPC6–PdCl6 aggregates (orange: C; blue: N; green: Cl; dark green: Pd). | ||
According to the XRD patterns, the structural simulation suggests that the aggregates of PQPC6–PdCl6 have a similar packing model to that of PQPC6–PtCl4 with their anions embedded between the solvophilic interfaces (Fig. 4b). However, since the ionic radius of PdCl62− is larger than that of PtCl42−, the steric hindrance caused by the anions will push them away from the PQPC6+ cations. As a result, the packing of PQPC6–PdCl6 along the Y-axis is restricted since there is not enough space for the accommodation of PdCl62− to balance the increased positive charge density along the Y-axis. Thus, the width of the aggregates is shorter than that of the nanosheets from PQPC6–PtCl4, resulting in the ribbon-like morphology. Although different anions can cause varied morphologies, our experimental results indicate that the combination of PtCl42− and PdCl62− won't lead to the formation of aggregates with a new morphology and only the mixture of nanosheets and nanoribbons can be obtained (Fig. S3†).
We further explored the impact of the counteranions with different charges on the PQP-based ionic complexes; the self-assembly behavior of PQPC6–AuCl4 was thus investigated in this work. Interestingly, only rod-like aggregates with a length in the range from 600 nm to 1 μm and a diameter ranging from 60 to 100 nm were obtained (Fig. 5a and b). AFM characterization (Fig. 5c) reveals a thickness of ∼67 nm. Different from PQPC6–PtCl4 and PQPC6–PdCl6, the WAXD patterns of PQPC6–AuCl4 contain four sharp diffraction peaks at 6.32, 8.36, 9.60, and 12.74°, corresponding to the (001), (010), (100), (002) facets of the triclinic structure. The simulation profile of PQPC6–AuCl4 (Fig. 5d) also fits well with the experimental data with wRp and Rp converged to 11.74 and 8.63% and the parameters of the unit cell for PQPC6–AuCl4 are determined to be a = 10.63 Å, b = 10.67 Å, and c = 16.00 Å.
 | ||
| Fig. 5 (a) SEM and (b)TEM images; (c)AFM images of 1D rod formed by ionic complex PQPC6–AuCl4; (d) the experimental WAXS pattern (black) and the Pawley refined one (red) of PQPC6–AuCl4; (e) the theoretical possible 2D staggered structure of the PQPC6–AuCl4 aggregates (blue: C; orange: N; green: Cl; dark green: Au). | ||
According to the above calculation, the simulated packing model of PQPC6–AuCl4 is shown in Fig. 5e. Compared with PQPC6–PtCl4, the electrostatic repulsion between the adjacent dimers of PQPC6–AuCl4 along the Y-axis would increase significantly since AuCl4− is a monovalent anion, which would remarkably restrict the growth rate along the Y-axis. Thus, the 1D rod-like aggregates are derived from PQPC6–AuCl4.
In conclusion, we have demonstrated the synthesis and self-assembly of three hexyl-substituted PQP salts with different inorganic anions. By using counter-anions with different sizes and valances, the packing of PQPC6 cations along different dimensions could be easily tuneable, giving rise to different morphologies from 2D nanosheets to 1D nanorods. In line with these arguments, it could be concluded that the divalent inorganic anions (PtCl42− and PdCl62−) in the PQPC6-based ionic complexes can effectively adjust the non-covalent interactions between the adjacent PQPC6 cations and direct the packing of the PQPC6 salts into 2D aggregates. Thus, it is anticipated that this approach can be applied to restructure the order and arrangement of the functional organic molecules in their aggregates, which will provide new opportunities for the fabrication of miniaturized optoelectronic devices.
Acknowledgements
This work was financially supported by the 973 Program of China (2014CB239701, 2013CBA01602 and 2012CB933404), Natural Science Foundation of China (21320102006, 61235007, 61575121, 21572132 and 21372155), Professor of Special Appointment at Shanghai Institutions of Higher Learning, Science and Technology Commission of Shanghai Municipal (12JC1404900), MPI-SJTU Partner Group Project (M.CH.A.Poly0004), ERC project on 2DMATER and EU Graphene Flagship. We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for the characterization of the materials.Notes and references
- (a) X. Huang, C. Tan, Z. Yin and H. Zhang, Adv. Mater., 2014, 26, 2185 CrossRef CAS; (b) R. Mas-Balleste, C. Gomez-Navarro, J. Gomez-Herrero and F. Zamora, Nanoscale, 2011, 3, 20 RSC; (c) C. N. R. Rao, H. S. S. Ramakrishna Matte and U. Maitra, Angew. Chem., Int. Ed., 2013, 52, 13162 CrossRef CAS; (d) J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schluter, Angew. Chem., Int. Ed., 2009, 48, 1030 CrossRef CAS.
- (a) M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef; (b) T. Govindaraju and M. B. Avinash, Nanoscale, 2012, 4, 6102 RSC.
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403 CrossRef CAS.
- (a) T. Bauer, Z. Zheng, A. Renn, R. Enning, A. Stemmer, J. Sakamoto and A. D. Schlüter, Angew. Chem., 2011, 123, 8025 CrossRef; (b) Y. Zheng, H. Zhou, D. Liu, G. Floudas, M. Wagner, K. Koynov, M. Mezger, H.-J. Butt and T. Ikeda, Angew. Chem., 2013, 125, 4945 CrossRef.
- W. Yao, Y. Yan, L. Xue, C. Zhang, G. Li, Q. Zheng, Y. S. Zhao, H. Jiang and J. Yao, Angew. Chem., 2013, 125, 8875 CrossRef.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931 RSC.
- E. J. Robertson, G. K. Olivier, M. Qian, C. Proulx, R. N. Zuckermann and G. L. Richmond, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 338 CrossRef.
- (a) D. Wu, X. Feng, M. Takase, M. C. Haberecht and K. Müllen, Tetrahedron, 2008, 64, 11379 CrossRef CAS; (b) D. Wu, W. Pisula, V. Enkelmann, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 9620 CrossRef CAS; (c) D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686 CrossRef CAS; (d) D. Wu, L. Zhi, G. J. Bodwell, G. Cui, N. Tsao and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5417 CrossRef CAS.
- D. Wu, R. Liu, W. Pisula, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2791 CrossRef CAS.
- J. Zhang, Y. Yan, M. W. Chance, J. Chen, J. Hayat, S. Ma and C. Tang, Angew. Chem., 2013, 125, 13629 CrossRef.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672 CrossRef CAS.
- S. Ghosh, A. Roy, D. Banik, N. Kundu, J. Kuchlyan, A. Dhir and N. Sarkar, Langmuir, 2015, 31, 2 Search PubMed.
- W. M. Haynes, T. J. Bruno and D. R. Lide, CRC Handbook of Chemistry and Physics, Internet Version, 85th edn, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis methods and additional characterization are presented. See DOI: 10.1039/c5ce02171e |
| This journal is © The Royal Society of Chemistry 2016 |