DOI:
10.1039/C5CE02156A
(Paper)
CrystEngComm, 2016,
18, 3574-3584
A detailed comparative structural study of the hydrogen bonded networks in solids, obtained by the reaction of 4,4′-bipyridine and varied alkane-α,ω-diphosphonic acids†
Received
3rd November 2015
, Accepted 9th April 2016
First published on 29th April 2016
Abstract
In a comparative structural study, the solid state structures of seven new compounds obtained by the reaction of 4,4′-bipyridine and alkane-α,ω-diphosphonic acids with different chain lengths have been characterised by single crystal X-ray diffraction and vibrational spectroscopy. The compounds are composed of either [4,4′-Hbipy]+ or [4,4′-H2bipy]2+ cations and [H2O3P(CH2)nPO3H]− anions and exhibit layered structures with clearly separated anionic and cationic areas. Within the three [4,4′-Hbipy]+ salts, the 4,4′-bipyridin-1-ium cations are connected to adjacent ones by medium strong, charge supported N+–H⋯N hydrogen bonds to form infinite chains. The anionic substructures of these compounds are quite different. The crystal structures of the five [4,4′-H2bipy]2+ salts consist of anionic, hydrogen bonded strands which are linked to adjacent ones by 4,4′-bipyridine-1,1′-diium cations. All but one can be broken down into two different types of structures in which the cations either lie in line with the anionic strands (type 1) or perpendicular to the aforementioned (type 2). For [4,4′-H2bipy][H2O3P(CH2)6PO3H]2 (2), two concomitant polymorphs have been obtained and characterised by single crystal and powder X-ray diffraction. Both polymorphs were classifiable into type 1 and 2, respectively. Only [4,4′-H2bipy][H2O3P(CH2)9PO3H]2 (4) is a three-dimensional hydrogen bonded framework and therefore not assignable to one of these two types of structures. π-System distances and a remarkable bending of the alkylene chains document the contribution of π–π-stacking and van der Waals interactions to the lattice enthalpy.
Introduction
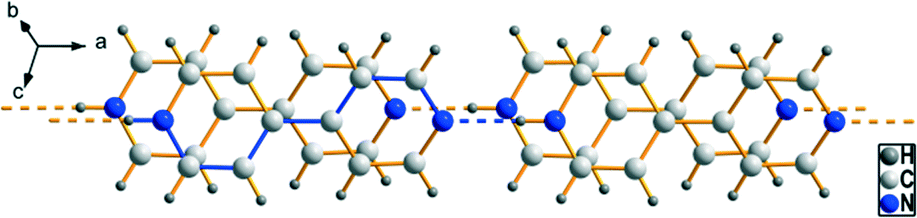
Materials constructed from organophosphonic acids have attracted much attention in the fields of crystal engineering not only because of their interesting topologies and structural diversity but also due to their diverse functional properties. For instance, metal phosphonates exhibit a low solubility and resistance against chemicals. They have potential applications regarding catalysis,1–4 ion exchange,5–7 proton conductivity,8–11 intercalation chemistry,12–15 photochemistry16–22 and materials chemistry.23–26 Oligofunctional derivatives like aminophosphonic acids, carboxyphosphonic acids and diphosphonic acids are promising tectons for the preparation of pillared layered metal phosphonates and the formation of microporous solids.25,27–32 In 1983, Dines et al. were the first to use diphosphonate tectons for the construction of microporous materials.33 Although metal phosphonates synthesised from these oligofunctional phosphonic acids are increasingly applied as organic–inorganic hybrid materials in materials chemistry, their structural elucidation is often handicapped by the lack of crystalline material suitable for powder or single crystal X-ray diffraction studies.34 However, organic salts of organophosphonic acids, e.g. with primary,35–39 secondary40–42 and tertiary39,43–48 amines, seem to be feasible model compounds for metal phosphonates as they exhibit similar structural characteristics and are less complicated to crystallise. A common feature of the synthesis of these materials in solution is the transfer of a proton from the phosphonic acid to the amine. Furthermore, the resulting [R–PO2(OH)]− anions are able to act as both hydrogen bond donor and hydrogen bond acceptor. Thus, these organic solids establish extended hydrogen bonded networks which include a rich diversity of strong, charge supported hydrogen bonds and can either be one-, two- or three-dimensional.40 Also protonated long-chained diphosphinic acid tectons have been used to construct hydrogen bonded frameworks.49 Investigations of the solid state structures of alkane-α,ω-diphosphonic acids, (HO)2OP–(CH2)n–PO(OH)2 (n = 1–12), have shown that the geometric parameters of the phosphonic acid moiety as well as its tridentate character benefit the formation of three-dimensional hydrogen bonded architectures.38,50–53 In all structures of the alkane-α,ω-diphosphonic acids, each P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen atom acts as an acceptor for two hydrogen bonds and each P–OH group participates in a single hydrogen bond as a donor. In terms of graph set analysis,54–57 R22(8), which is formed by a dimeric unit of phosphonic acid groups, and R24 (2(n + 1) + 12), whose ring size depends on the chain length n of the corresponding diphosphonic acid, are the most frequently appearing hydrogen bonded ring motifs (Fig. 1). The hydrogen bonded chain motif C(4) [⋯OP–O–H⋯]n can also be found in the majority of the crystal structures of the alkane-α,ω-diphosphonic acids. It is conceivable that these structural characteristics are retained e.g. within aminium salts of alkane-α,ω-diphosphonic acids and can be utilised to design new organic solids with predictable structures. Especially, 4,4′-bipyridine seems to be a suitable crystal engineering tecton for the creation of hydrogen bonded networks.44,47 In this work, we report the crystal structures and vibrational spectra of eight novel hydrogen bonded networks in the 4,4′-bipyridine/alkane-α,ω-diphosphonic acid system as part of our continuing interest in general concepts within the fields of crystal engineering.
O oxygen atom acts as an acceptor for two hydrogen bonds and each P–OH group participates in a single hydrogen bond as a donor. In terms of graph set analysis,54–57 R22(8), which is formed by a dimeric unit of phosphonic acid groups, and R24 (2(n + 1) + 12), whose ring size depends on the chain length n of the corresponding diphosphonic acid, are the most frequently appearing hydrogen bonded ring motifs (Fig. 1). The hydrogen bonded chain motif C(4) [⋯OP–O–H⋯]n can also be found in the majority of the crystal structures of the alkane-α,ω-diphosphonic acids. It is conceivable that these structural characteristics are retained e.g. within aminium salts of alkane-α,ω-diphosphonic acids and can be utilised to design new organic solids with predictable structures. Especially, 4,4′-bipyridine seems to be a suitable crystal engineering tecton for the creation of hydrogen bonded networks.44,47 In this work, we report the crystal structures and vibrational spectra of eight novel hydrogen bonded networks in the 4,4′-bipyridine/alkane-α,ω-diphosphonic acid system as part of our continuing interest in general concepts within the fields of crystal engineering.
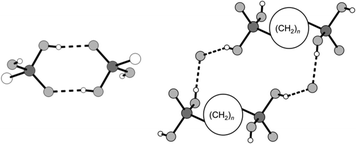 |
| | Fig. 1 Schematic illustration of the hydrogen bonded motifs R22(8) (left) and R24 (2(n + 1) + 12) (right) occurring in most of the alkane-α,ω-diphosphonic acids' crystal structures.53 | |
Experimental section
Synthesis and characterisation
The alkane-α,ω-diphosphonic acids were synthesised according to the literature.58–60 All other chemicals were obtained from commercial sources and used as purchased. The title compounds [4,4′-Hbipy][H2O3P(CH2)3PO3H] (1), [4,4′-H2bipy][H2O3P(CH2)6PO3H]2 (2a) and (2b), [4,4′-H2bipy][H2O3P(CH2)8PO3H]2 (3), [4,4′-H2bipy][H2O3P(CH2)9PO3H]2 (4), [4,4′-H2bipy][H2O3P(CH2)10PO3H]2 (5), [4,4′-H2bipy][H2O3P(CH2)11PO3H]2 (6), [4,4′-Hbipy][H2O3P(CH2)12PO3H]·H2O3P(CH2)12PO3H2 (7) were prepared by dissolving equimolar quantities (0.5 mmol) of 4,4′-bipyridine and the corresponding alkane-α,ω-diphosphonic acid in methanol separately. The solutions were mixed and set aside exposed to air. Within several days, colourless platelet-like crystals suitable for single crystal X-ray diffraction were obtained in all cases. The single crystals of compounds 1–7 were mounted on an Oxford Xcalibur four-circle diffractometer61 equipped with an EOS detector and a gas stream cooling device. X-ray powder diffraction patterns were measured with a Huber G600 Guinier diffractometer working in transmission mode. The utilised Cu Kα1 radiation was generated using a quartz (101) monochromator. Comparative powder patterns were simulated from the single crystal data with the PowderCell 2.3 programme.62 IR spectra were recorded on a Digilab Excalibur FTS 3500 spectrometer63 with an apodised resolution of 4 cm−1 using a MIRacle ATR unit64 in the region between 4000 and 530 cm−1. Raman spectra were measured on a MultiRAM spectrometer65 equipped with a Nd-YAG laser (1064 nm) and an InGaAs detector (resolution: 8 cm−1; range: 4000–70 cm−1). Elemental analyses (C, H, N) were performed with a HEKAtech Euro EA3000 instrument.66 Differential scanning calorimetry experiments were carried out with a Mettler Toledo DSC 1 calorimeter67 equipped with a FRS 5 sensor using aluminium crucibles with perforated caps. Melting points were determined using the Mettler Toledo melting point system MP90. The results of the IR and Raman spectroscopic measurements as well as the results of elemental analysis and melting point determinations are deposited in the ESI† file.
Crystal structure determinations
Data collection and data reduction followed the standard routine ensuring a completeness of more than 99% of reflections necessary for the Laue class in each case. Absorption corrections were applied using the multi-scan method. Structure solutions by Direct Methods68 generally yielded all non-hydrogen atom positions. Difference Fourier maps and simultaneous successive refinement of the primary structure solutions gave almost completed models. In the latest stages of refinement anisotropic displacement parameters for all non-hydrogen atoms were refined. Hydrogen atoms were included using suitable riding models of the SHELX programme system:69 AFIX 23 for methylene groups, AFIX 43 for methine and aminium groups and AFIX 147 for hydroxyl groups. Selected crystallographic data are given in Table 1. CCDC 1433066 (1), 1004560 (2a), 1433181 (2b), 1433067 (3), 1433068 (4), 1433279 (5), 1433280 (6) and 1433281 (7) contain the supplementary crystallographic data for this paper.
Table 1 Crystallographic data and details of the structure refinement for 1–7
| Compound |
1
|
2a
|
2b
|
3
|
4
|
5
|
6
|
7
|
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo|.
wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}0.5.
|
| Empirical formula |
C13H18N2O6P2 |
C22H40N2O12P4 |
C22H40N2O12P4 |
C26H48N2O12P4 |
C28H52N2O12P4 |
C30H56N2O12P4 |
C32H60N2O12P4 |
C34H64N2O12P4 |
|
M
r [g mol−1] |
360.2 |
648.4 |
648.4 |
704.5 |
732.6 |
760.6 |
788.7 |
816.8 |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P |
Pc
|
P21/c |
P |
P |
|
a [Å] |
9.8401(4) |
19.5429(8) |
9.1687(4) |
9.1052(2) |
42.860(9) |
23.4234(7) |
4.75510(10) |
9.1869(18) |
|
b [Å] |
12.0388(5) |
4.71970(10) |
9.6050(4) |
9.5324(3) |
11.426(2) |
4.64205(13) |
8.7442(3) |
9.2725(19) |
|
c [Å] |
15.2173(5) |
17.2626(7) |
17.9827(7) |
20.3596(6) |
7.3826(15) |
16.9643(5) |
22.6864(8) |
24.348(5) |
|
α [°] |
95.386(3) |
90 |
79.130(3) |
79.976(2) |
90 |
90 |
81.639(3) |
83.68(3) |
|
β [°] |
106.844(3) |
114.668(5) |
89.668(3) |
86.857(2) |
90.59(3) |
104.720(3) |
87.130(3) |
81.01(3) |
|
γ [°] |
112.289(4) |
90 |
70.320(3) |
69.836(2) |
90 |
90 |
88.196(3) |
81.40(3) |
|
V [Å3] |
1553.60(11) |
1446.93(10) |
1461.54(11) |
1633.45(8) |
3615.2(12) |
1784.02(10) |
931.81(5) |
2017.6(7) |
|
Z
|
4 |
2 |
2 |
2 |
4 |
2 |
2 |
2 |
|
D
calcd. [g cm−3] |
1.54 |
1.49 |
1.47 |
1.43 |
1.35 |
1.42 |
1.41 |
1.34 |
|
μ [mm−1] |
0.313 |
0.324 |
0.321 |
0.294 |
0.268 |
0.275 |
0.266 |
0.248 |
|
T
min/Tmax |
0.939/1.000 |
0.885/1.000 |
0.920/1.000 |
0.902/1.000 |
0.244/0.267 |
0.831/1.000 |
0.960/1.000 |
0.931/1.000 |
|
F(000) |
752 |
684 |
684 |
748 |
1560 |
812 |
422 |
876 |
|
T [K] |
100 |
294 |
294 |
105 |
173 |
106 |
122 |
110 |
|
λ [Å] |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| 2ϑmax [°] |
54.99 |
50.00 |
50.00 |
50.00 |
53.83 |
50.00 |
50.00 |
50.00 |
| Completeness [%] |
99.8 |
99.8 |
99.8 |
99.8 |
97.8 |
99.8 |
98.7 |
99.8 |
| Total reflections |
19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 960 960 |
19524 |
15954 |
27285 |
26931 |
20584 |
7612 |
21099 |
| Independent reflections |
7121 |
2563 |
5133 |
5745 |
12351 |
3141 |
3257 |
7072 |
|
R
int
|
0.0386 |
0.0276 |
0.0268 |
0.0345 |
0.0399 |
0.0812 |
0.0285 |
0.0380 |
| Parameters ref./restr. |
422/0 |
190/0 |
367/0 |
403/0 |
841/2 |
220/0 |
229/0 |
476/0 |
|
R
1
(I > 2σ(I)) |
0.0406 |
0.0334 |
0.0340 |
0.0297 |
0.0504 |
0.0435 |
0.0375 |
0.0717 |
| wR2b (all data) |
0.0919 |
0.0691 |
0.0930 |
0.0912 |
0.1079 |
0.0974 |
0.0967 |
0.1933 |
| Goodness of fit |
1.042 |
1.305 |
1.049 |
1.046 |
1.132 |
1.019 |
1.048 |
1.115 |
| Δρmin/max [e Å−3] |
−0.408/0.431 |
−0.330/0.351 |
−0.397/0.390 |
−0.369/0.416 |
−0.266/0.332 |
−0.368/0.388 |
−0.372/0.425 |
−0.540/0.996 |
Results and discussion
[4,4′-Hbipy]+ salts of alkane-α,ω-diphosphonic acids
All yet structurally characterised [4,4′-Hbipy]+ salts of alkane-α,ω-diphosphonic acids comprise 4,4′-bipyridin-1-ium cations linked to adjacent ones by medium strong, charge supported N+–H⋯N hydrogen bonds to form infinite chains (Fig. 2) which can be described by the symbol C(9) in terms of graph-set analysis.54–57
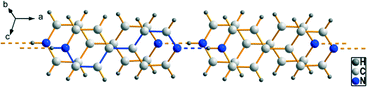 |
| | Fig. 2 Pairwise π–π-stacking of infinite hydrogen bonded [4,4′-Hbipy]+ chains within the crystal structure of 1. Showing the parallel displaced face-to-face arrangement as well as the graph-set C(9) indicated by blue bonds. | |
D⋯A distances between 2.591(3) and 2.815(2) Å and bond angles of 176–180° are observed. Additionally, these pillared cationic chains are stacked in parallel displaced face-to-face arrangements either pairwise or in layers consisting of multiple chains.70 The perpendicular distances between the planes of neighbouring pyridinium rings within these π–π-stackings are in the range between 3.40 and 3.62 Å and the centroid offsets amount to 1.34–3.07 Å. These values are representative for interactions at medium strength.70 Intramolecular bond lengths and angles of the 4,4′-bipyridin-1-ium cations are within the expected ranges. Moreover, the pyridinium moieties of the [4,4′-Hbipy]+ cations are oriented coplanar or twisted against each other up to an angle of torsion of 27.1(3)° depending on the packing conditions and their positioning within the anionic substructures. Although the cationic substructures within the here presented [4,4′-Hbipy]+ salts are basically very similar, the anionic substructures exhibit some fundamental differences.
[H2O3P(CH2)3PO3H]−.
The anionic substructure of 1 consists of hydrogen (3-phosphonopropyl)phosphonate ions which establish head-to-tail connected dimeric units via medium strong O–H⋯O hydrogen bonding (Fig. 3). Resulting hydrogen bonded motifs are a 16-membered ring, which is classified as a first level graph-set R22(16), and a 8-membered ring, classified as a second level graph-set R22(8). Additionally, the hydrogen (3-phosphonopropyl)phosphonate anions are connected to neighbouring dimeric units by strong, charge supported O–H⋯O− hydrogen bonds forming infinite strands running parallel to the cationic chains along the a axis (Fig. 3). In this case, the resulting hydrogen bonded motif is a 16-membered ring, classified as a second level graph-set R44(16). The D⋯A distances for the hydrogen bonds within the anionic substructure lie between 2.4048(19) and 2.542(2) Å and the bond angles amount to 158–171°. Intramolecular bond lengths and angles of the hydrogen (3-phosphonopropyl)phosphonate anions are within the expected ranges and comparable with those found in the structure of propane-1,3-diphosphonic acid.51 Within the PO3H− group there is a significant difference in the P–O bond lengths between the P–OH bond and those in the PO2− fragment. Furthermore the O–P–O angle of the PO2− fragment is considerably larger than tetrahedral.44 In the structure of 1, the pillared anionic and cationic substructures are packed alternating along [011] held together by comparatively strong π–π-interactions as well as Coulomb and van der Waals forces (Fig. 4, upper part).
 |
| | Fig. 3 The anionic substructure of 1 exhibits dimeric hydrogen bonded units of [H2O3P(CH2)3PO3H]− ions which are further connected to infinite strands. Corresponding graph-set descriptors R22(8), R44(16) and R22(16) are indicated by red, green and blue bonds, respectively. A corresponding constructor graph is shown below.56,57 | |
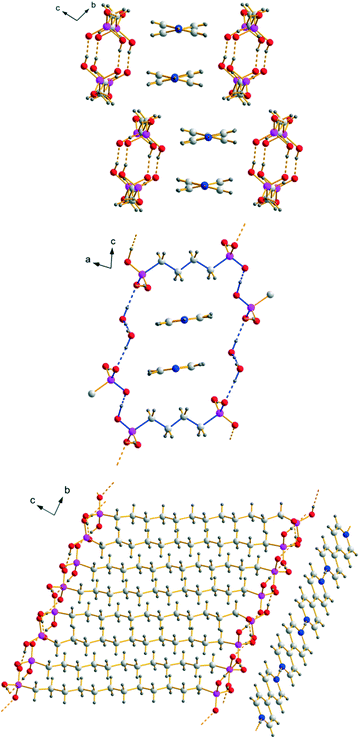 |
| | Fig. 4 Packing diagrams of the anionic and cationic substructures within the three [4,4′-Hbipy]+ containing hydrogen bonded networks: [4,4′-Hbipy][H2O3P(CH2)3PO3H] (1) (top), [4,4′-Hbipy][H2O3P(CH2)4PO3H]·2H2O (middle) and [4,4′-Hbipy][H2O3P(CH2)12PO3H)]·H2O3P(CH2)12PO3H2 (7) (bottom) (R88(34) is indicated by blue bonds). | |
[H2O3P(CH2)4PO3H]−·2H2O (ref. 47).
Within the anionic substructure of [4,4′-Hbipy][H2O3P(CH2)4PO3H]·2H2O known from the literature, the hydrogen (4-phosphonobutyl)phosphonate ions form two-dimensional hydrogen bonded networks in the ab plane (Fig. 5). The strong to medium strong O–H⋯O hydrogen bonds exhibit D⋯A distances from 2.463(3) to 2.624(3) Å and bond angles ranging from 166(4) to 178(3)°. Resulting hydrogen bonded motifs are a 16-membered ring R44(16) and a 18-membered ring R22(18), which is comparable to R22(16) in the structure of 1, as well as the chain motif C22(8), which is similarly found within the crystal structures of several alkane-α,ω-diphosphonic acids. The intramolecular bond lengths and angles within the hydrogen(4-phosphonobutyl)phosphonate anions are unexceptional and comparable with the findings described for the anions in 1. Along the c axis, the two-dimensional anionic networks are further connected by the water molecules to form a three-dimensional hydrogen bonded framework which exhibits rectangular channels with cavities of approximately 11.0 × 7.9 Å (Fig. 4, middle part). These channels, which can be described with the symbol R88(34) in terms of graph-set analysis, are occupied by pairwise π–π-stacked infinite [4,4′-Hbipy]+ chains running along the b axis. Structural data of [4,4′-Hbipy][H2O3P(CH2)4PO3H]·2H2O can be obtained from The Cambridge Crystallographic Data Centre via the deposition number 208719 (CSD entry: OBOZIP).
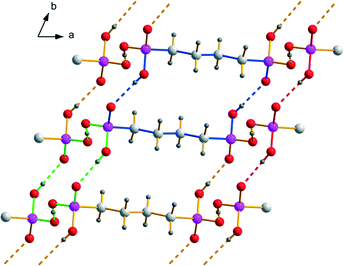 |
| | Fig. 5 Hydrogen bonding within the two-dimensional networks composed of [H2O3P(CH2)4PO3H]− ions with R22(18) (blue), R44(16) (green) and C22(8) (red). | |
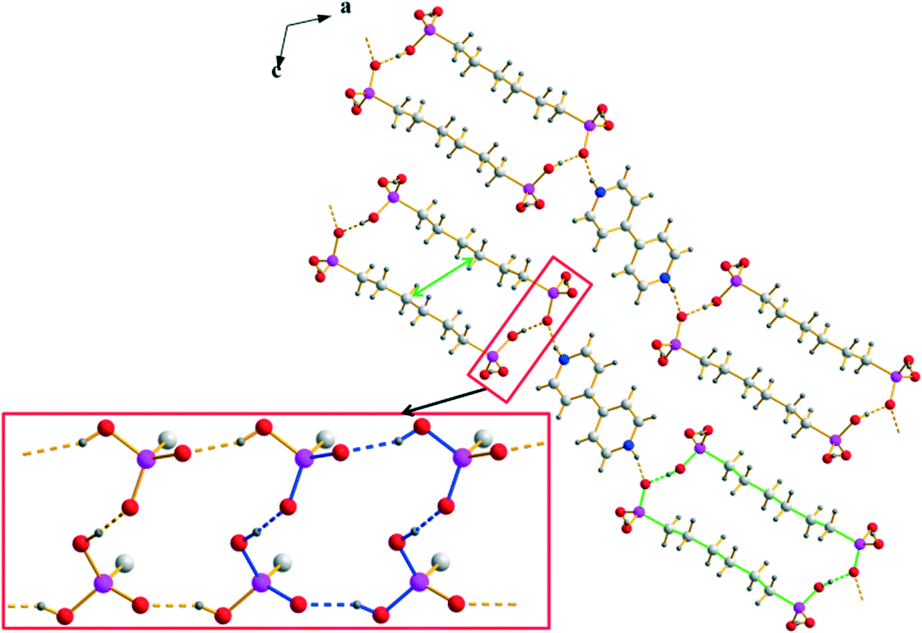
[H2O3P(CH2)12PO3H)]−·H2O3P(CH2)12PO3H2.
The anionic substructure of 7 contains two-dimensional hydrogen bonded networks in the ab plane (Fig. 6) which are pairwise connected with each other by alkylene chains along the c axis to form a typical bilayer with a hydrophobic interior and a hydrophilic exterior (Fig. 4, lower part). The corresponding D⋯A distances of the strong to medium strong O–H⋯O hydrogen bonds lie between 2.435(4) and 2.674(4) Å and the bond angles amount to 154(2)–176(2)°. Within the two-dimensional networks, three different hydrogen bonded ring motifs can be observed, classifiable as the fourth level graph-sets R34(14) and R44(16) as well as the fifth level graph-set R56(22). Within the hydrophobic regions of the anionic substructure, neighbouring alkylene chains interact with each other through van der Waals forces which are of comparable strength as in the related n-alkanes.71 The intramolecular bond lengths and angles within the hydrogen (12-phosphonododecyl)phosphonate anions and the dodecane-1,12-diphosphonic acid molecules are unexceptional and lie within the expected ranges. In the structure of 7, the anionic and cationic substructures are arranged in layers alternating along the c axis held together by Coulomb as well as van der Waals forces (Fig. 4). Contrary to the structure of 1 in which the pillared anionic and cationic substructures run parallel to each other along the a axis, the moieties within the anionic substructure of 7 are arranged with its alkylene chains approximately perpendicular to the propagation direction of neighbouring [4,4′-Hbipy]+ chains.
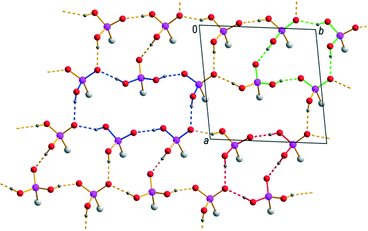 |
| | Fig. 6 Two-dimensional hydrogen bonded networks within the anionic substructure of 7 with the corresponding graph-set descriptors R34(14) (red), R44(16) (green) and R56(22) (blue). | |
[4,4′-H2bipy]2+ salts of alkane-α,ω-diphosphonic acids
Most of the [4,4′-H2bipy]2+ salts of alkane-α,ω-diphosphonic acids exhibit layered crystal structures in which two-dimensional hydrogen bonded strands consisting of hydrogen (ω-phosphonoalkyl)phosphonate anions are linked by 4,4′-bipyridine-1,1′-diium cations via medium strong, charge supported N+–H⋯O hydrogen bonds along one direction (Fig. 7, 8 and 9). The corresponding D⋯A distances lie between 2.521(6) and 2.676(2) Å and the bond angles amount to 164.4–175.1°. Similar to the structures of the corresponding [4,4′-Hbipy]+ containing salts, the cations within the [4,4′-H2bipy]2+ salts reported here are π–π-stacked in parallel displaced face-to-face arrangements with perpendicular distances between neighbouring pyridinium rings of 3.30–3.62 Å and centroid offsets ranging between 0.85 and 1.58 Å. These values represent strong to medium strong interactions. The vast majority of the [4,4′-H2bipy]2+ cations within the structures discussed here exhibit an almost coplanar conformation of the two pyridinium moieties with a maximum angle of torsion of 2.9(3)° concerning the twist of the two pyridinium rings. All other intramolecular bond lengths and angles of the cations are within the expected ranges.72,73 The linkage of the [4,4′-H2bipy]2+ cations and the anionic strands of hydrogen (ω-phosphonoalkyl)phosphonates results in two-dimensional hydrogen bonded frameworks which are stacked in layers and interact with neighbouring frameworks through Coulomb as well as van der Waals forces. The hydrogen bonded networks described afterwards can mainly be classified into two different types of structures according to the arrangement of their cationic and anionic parts.
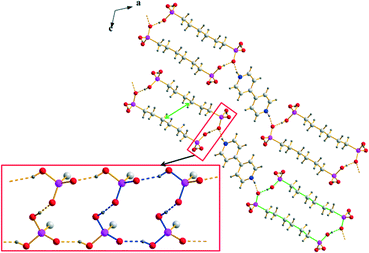 |
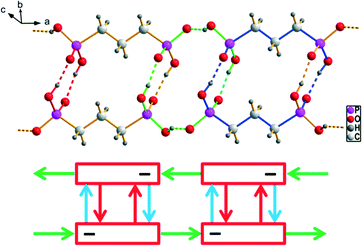
| | Fig. 7 Packing diagram of 2a exemplary for type 1 structures with the linearly arranged two-dimensional hydrogen bonded frameworks consisting of [4,4′-H2bipy]2+ cations and [H2O3P(CH2)6PO3H]− anions. The hydrogen bonding within the anionic strands is shown in the red box (R22(22) and R44(16) are indicated by green and blue bonds, respectively). The green arrow shows the shortest van der Waals contact. | |
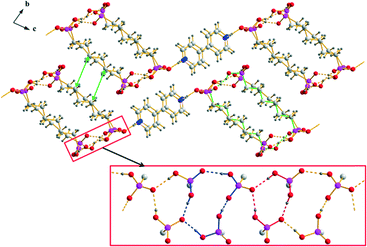 |
| | Fig. 8 Packing diagram of 2b (similar for 3) with the hydrogen bonded [4,4′-H2bipy]2+ cations and [H2O3P(CH2)6PO3H]− anions arranged in a herringbone pattern. The hydrogen bonded ribbons within the anionic substructures of 2b and 3 are shown in the red box (R22(22), R23(10) and R34(14) are indicated by green, red and blue bonds, respectively). The green arrows show the shortest van der Waals contacts. | |
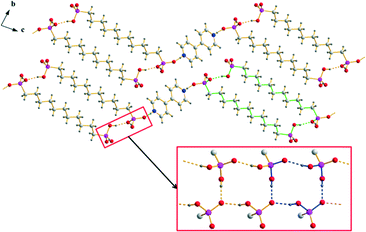 |
| | Fig. 9 Packing diagram of 6 with the hydrogen bonded [4,4′-H2bipy]2+ cations and [H2O3P(CH2)11PO3H]− anions arranged in a herringbone pattern. The hydrogen bonding within the anionic strands of 6 is shown in the red box (R22(32) and R34(14) are indicated by green and blue bonds, respectively). | |
Type 1.
The anionic substructures within the homotypic hydrogen bonded networks of 2a and 5 contain hydrogen bonded ribbons which are formed by –PO3H2 and –PO3H− groups and run along the b axis (Fig. 7). The corresponding D⋯A distances of the medium strong O–H⋯O hydrogen bonds amount to 2.512(2)–2.563(2) Å and the bond angles lie between 157.0 and 173.6°. Within these ribbons, a hydrogen bonded 16-membered ring motif occurs which is classified as a third level graph-set R44(16). Moreover, the connection of respectively two hydrogen bonded ribbons along [101] via the alkylene chains of the anions results in the ring motifs R22(22) for 2a and R22(30) for 5. The hydrogen (ω-phosphonoalkyl)phosphonates in the anionic substructures exhibit almost all-transoid conformations with bond lengths and angles within the expected ranges. However, their alkylene chains are slightly bent towards the inside of the hydrogen bonded strands resulting in considerably shorter van der Waals contacts with a C⋯C distance of 3.96 Å in the middle (see green arrows in Fig. 7). Along [101], the anionic hydrogen bonded strands in the structures of type 1 are linked with each other by roughly parallel arranged, π–π-stacked [4,4′-H2bipy]2+ cations to form two-dimensional frameworks.
Type 2.
The anionic substructures within the homotypic hydrogen bonded networks 2b and 3 consist of strands in which respectively two hydrogen bonded ribbons are linked by the alkylene chains of the hydrogen (ω-phosphonoalkyl)phosphonate anions. However, the connection of the –PO3H2 and –PO3H− groups within these hydrogen bonded ribbons is completely different from that in the structures of type 1. The hydrogen bonded motifs within the ribbons which run along the a axis are a 10-membered ring classifiable as a third level graph-set R23(10) and a 14-membered ring classifiable as the fourth level graph-set R34(14) (Fig. 8). The corresponding D⋯A distances of the strong to medium strong O–H⋯O hydrogen bonds lie between 2.4595(16) and 2.619(2) Å and the bond angles amount to 154.4–172.3°. Similar to the structures of type 1, the connection of two neighbouring hydrogen bonded ribbons via the alkylene chains of the hydrogen (ω-phosphonoalkyl)phosphonate anions which exhibit slightly distorted all-transoid conformations results in the ring motifs R22(22) for 2b and R22(26) for 3. In this case, the alkylene chains are bent towards the outside of the hydrogen bonded strands. Therefore, potential van der Waals interactions within the strands (shortest C⋯C distance 4.29 Å) are considerably weaker than in the structures of type 1. Furthermore, the shortest C⋯C distances of neighboring strands are with a value of 4.35 Å (green arrows in Fig. 8) in the range of weak van der Waals interactions. Approximately along [011], the anionic hydrogen bonded strands of 2b and 3 are further connected with each other by the π–π-stacked [4,4′-H2bipy]2+ cations (Fig. 8). The two-dimensional frameworks formed this way are arranged in a herringbone pattern. In the crystal structure of 6, the anionic and cationic domains of the hydrogen bonded network are arranged in a herringbone pattern as well. However, the arrangement of the hydrogen (11-phosphonoundecyl)phosphonates within the anionic substructures is more related to the structures of type 1 than to the other structures of type 2. The hydrogen bonded ribbons within the anionic substructures are composed of –PO3H2 and –PO3H− groups, run along the a axis and exhibit a 14-membered hydrogen bonded ring motif which is classifiable as the third level graph-set R34(14) (Fig. 9). The D⋯A distances of the corresponding O–H⋯O hydrogen bonds lie between 2.4911(19) and 2.5711(19) Å and the bond angles amount to 153.8–164.4°. Moreover, the connection of respectively two hydrogen bonded ribbons by the alkylene chains of the [H2O3P(CH2)11PO3H]− anions results in the hydrogen bonded ring motif R22(32). Here, the anions don’t show any form of bending and the C⋯C distances within the hydrogen bonded strands lie between 4.00 and 4.91 Å. Similar to the other structures of type 2, the anionic hydrogen bonded strands within the structure of 6 are linked by π–π-stacked [4,4′-H2bipy]2+ cations approximately along [011] to form two-dimensional frameworks (Fig. 9).
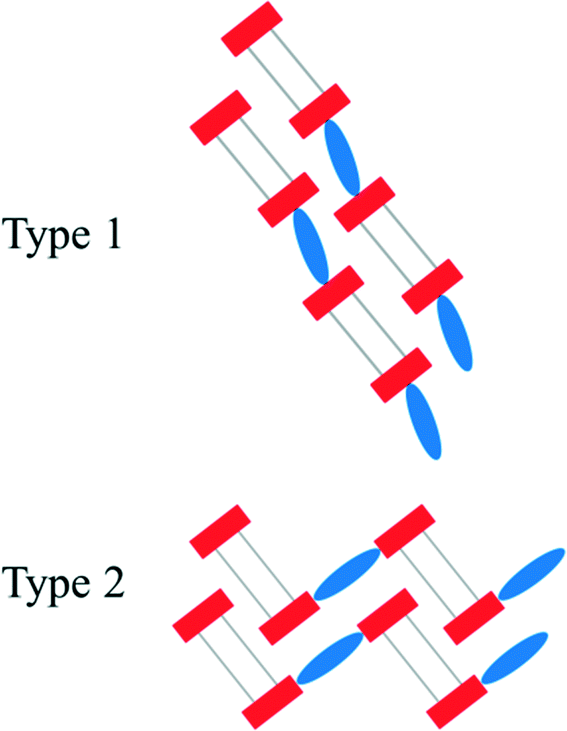
Comparative view.
In both types of structures, the stacking of the anionic, hydrogen bonded strands is nearly identical. However, the π–π-stacked [4,4′-H2bipy]2+ cations which connect these stacks can either lie in line with the anionic strands (type 1) or perpendicular to the aforementioned (type 2) (Fig. 10). The arrangement of type 2 results in a herringbone pattern. For [4,4′-H2bipy][H2O3P(CH2)6PO3H]2, a modification featuring the linear arrangement (2a) as well as a modification including the herringbone pattern (2b) was obtained. The polymorphism of 2 is described more in detail in a following section. Besides the hydrogen bonded network 2a, the homotypic structure of 5 also exhibits an in-line arrangement of the anionic and cationic domains. These structures represent type 1. On the other hand, the two homotypic compounds of 2b and 3 as well as compound 6 have their anionic and cationic domains adjusted in a herringbone pattern. The structure of [4,4′-H2bipy][HO3P(CH2)2PO3H]·2H2O (ref. 44) known from the literature shows a slightly different kind of herringbone pattern. These four structures are referred to as type 2.
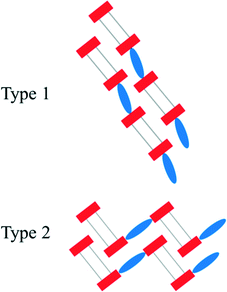 |
| | Fig. 10 Schematic representation of the two mainly occurring construction patterns within the [4,4′-H2bipy]2+ salts of alkane-α,ω-diphosphonic acids. The anionic strands consist of hydrogen bonded terminal groups (red rectangles) and alkylene chains (grey lines). The π–π-stacked [4,4′-H2bipy]2+ cations are drawn as blue ellipses. | |
[4,4′-H2bipy][H2O3P(CH2)9PO3H]2.
An exceptional case to the two mainly occurring structural arrangements is the structure of 4. Here, the [4,4′-H2bipy]2+ cations which connect the anionic substructures via medium strong, charge supported N+–H⋯O hydrogen bonds along [100] are oriented crosswise to each other (Fig. 11). However, the corresponding D⋯A distances with values between 2.521(6) and 2.556(7) Å (bond angles of 168.6–173.5°) are similar to those within the other [4,4′-H2bipy]2+ salts featured here. The π–π-stacking of the [4,4′-H2bipy]2+ cations in parallel displaced face-to-face arrangements is quite different as in this case only one pyridinium ring per dication is involved. The perpendicular distances between neighbouring pyridinium rings amount to 3.57–3.64 Å and the centroid offsets lie between 0.95 and 1.12 Å. Compared to the π–π-stacking parameters discussed before, these values denote slightly weaker interactions. Moreover, the two pyridinium rings of the [4,4′-H2bipy]2+ cations are considerably twisted against each other with torsion angles of 25.1(8)° and 27.0(8)°, respectively. The crystal structure of 4 exhibits two different anionic substructures which both consist of hydrogen bonded hydrogen (9-phosphonononyl)phosphonate ions. The first anionic substructure contains hydrogen bonded ribbons which are formed by –PO3H2 and –PO3H− groups and run along the c axis (Fig. 11, red box). The corresponding D⋯A distances of the strong to medium strong O–H⋯O hydrogen bonds amount to 2.426(6)–2.588(6) Å and the bond angles lie between 155.6 and 165.9°. Within these ribbons, hydrogen bonded 8-membered ring motifs classified as second level graph-sets R22(8) are connected with each other in a zig-zag-chain. The connection of respectively two hydrogen bonded ribbons approximately along the a axis via the alkylene chains of the anions results in two-dimensional frameworks. In contrast to the structures of the other [4,4′-H2bipy]2+ salts, the connection of two neighbouring hydrogen bonded ribbons via the alkylene chains in this substructure results in a further connection along the b axis (Fig. 11, left half). The second anionic substructure exhibits two-dimensional hydrogen bonded networks in the bc plane (Fig. 11, blue box) in which the –PO3H2 and –PO3H− groups are linked by strong to medium strong O–H⋯O hydrogen bonds with D⋯A distances between 2.456(5) and 2.580(5) Å and bond angles of 152.8–163.1°. Within these networks, hydrogen bonded 22-membered ring motifs can be found classifiable as third level graph-sets R56(22). Further hydrogen bonded ring motifs R22(28) occur from the connection of respectively two hydrogen bonded networks approximately along the a axis (Fig. 11, right half). The linkage of the [4,4′-H2bipy]2+ cations and the two different anionic substructure moieties results in a three-dimensional hydrogen bonded framework with clearly separated anionic and cationic layers.
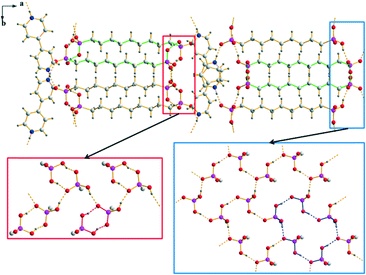 |
| | Fig. 11 Packing diagram of 4 with the crosswise stacking of the [4,4′-H2bipy]2+ cations and the two different anionic substructures consisting of [H2O3P(CH2)9PO3H]− anions. The hydrogen bonded ribbons within the first anionic substructure are shown in the red box and the hydrogen bonded two-dimensional networks within the second anionic substructure are shown in the blue box (R22(28), R22(8) and R56(22) are indicated by green, red and blue bonds, respectively). | |
Polymorphism of [4,4′-H2bipy][H2O3P(CH2)6PO3H]2.
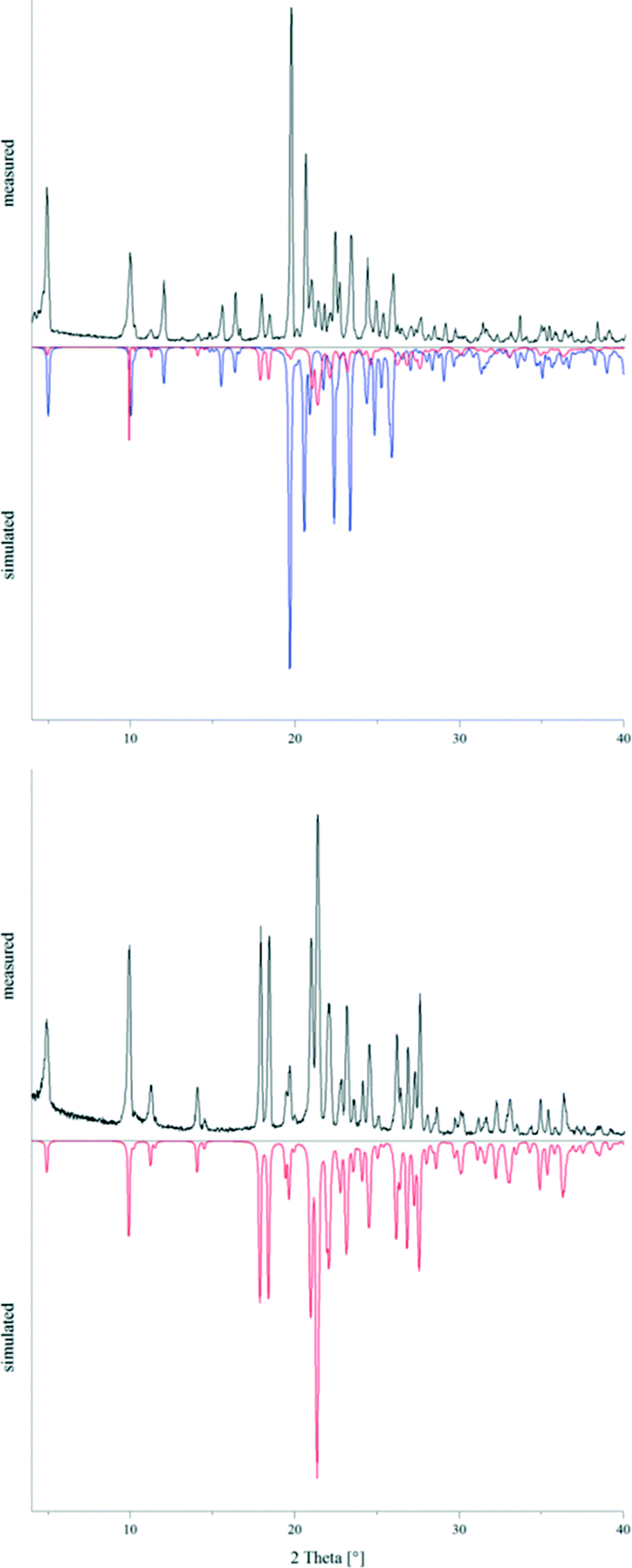
The powder pattern of a sample of 2, which was crystallised under ambient conditions, clearly showed the presence of the two concomitant polymorphs 2a and 2b in an approximate ratio of 1:9 (Fig. 12). After annealing parts of this powder sample in a sealed Duran glass ampoule in a block oven at 150 °C for five days, the recorded powder pattern revealed that 2a exclusively was present now. Subsequently, differential scanning calorimetry experiments were carried out for both powder samples in order to determine potential phase transformation and melting temperatures (also see the ESI† file). For the sample containing the two modifications 2a and 2b, two endothermic effects could be observed on heating (5 °C min−1) at Tpeak = 174.2 °C (Tonset = 154.0 °C) and Tpeak = 182.6 °C (Tonset = 176.6 °C), respectively. The DSC curve of 2a on the other hand only showed one endothermic effect on heating (5 °C min−1) at Tpeak = 182.3 °C (Tonset = 173.5 °C). Thus, the endothermic peak at 174.2 °C could be assigned to the phase transformation of 2b into 2a. The second endothermic peak at 182.6 °C reflected the melting temperature of 2a. In the region where lattice vibrations occur, the F-IR spectrum of 2a shows two bands, at 102 and 74 cm−1. The spectrum of the sample containing 2a and 2b exhibits three additional bands at 89, 54 and 36 cm−1 giving strong evidence for the presence of two different crystalline phases. Both F-IR spectra are depicted in the ESI† file.
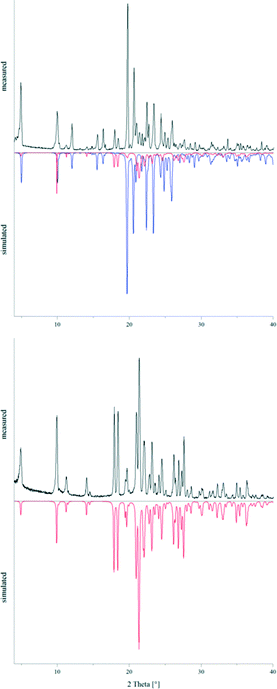 |
| | Fig. 12 Powder patterns of 2 before (top) and after (bottom) annealing at 150 °C for five days compared to the simulated powder patterns of 2a (red) and 2b (blue). | |
Stoichiometric aspects
At the beginning of this study all experiments were set up by using equimolar quantities of 4,4′-bipyridine and the corresponding alkane-α,ω-diphosphonic acids. Nevertheless, different compositions containing either mono- or diprotonated 4,4′-bipyridinium cations were obtained. In all cases, the alkane-α,ω-diphosphonic acid units were monodeprotonated. In order to further examine this finding we applied different stoichiometries of the educts. However, we solely obtained the same products already known from the reactions with equimolar quantities. The reaction product of decane-1,10-diphosphonic acid with a threefold molar excess of 4,4′-bipyridine has exemplary been characterised by powder X-ray diffraction. The corresponding pattern shown in the ESI† file reveals that the product which crystallised from the solution after two days was still [4,4′-H2bipy][H2O3P(CH2)10PO3H]2 (5) in pure form. The excess 4,4′-bipyridine crystallised several days later. So apparently, for some of these compounds the principles of packing outplay the stoichiometric conversion of the educts.
Vibrational spectroscopy
The IR spectra of the compounds 1–7 depicted in the ESI† file show the typical pattern for strongly hydrogen bonded systems which have multiple broad maxima associated with the O–H stretching vibration (2700, 2200 and 1600 cm−1).74–76 The relative intensities of these A, B and C type bands are quite similar with band C marginally stronger. Very often these bands have rather smooth contours or are even fused to a continuous region of enhanced absorption between 2900 and 1600 cm−1.74 Additionally, the IR and Raman spectra show the characteristic bands of the C–H stretching vibrations of the 4,4′-bipyridinium cations in the region of 3100–3000 cm−1 giving clear evidence for the presence of these protonated species in the solids. The characteristic pair of IR bands between 2960 and 2840 cm−1 whose intensity increases with an increasing chain length of the hydrogen (ω-phosphonoalkyl)phosphonate anion and which is in some cases split into several sub-bands can unambiguously be assigned to the asymmetric and symmetric C–H stretching vibrations of the methylene groups. However, the band of the N+–H stretching vibration which should appear between 2700 and 2250 cm−1 as a broad band seems to overlap with the aforementioned A and B type bands.77,78 Three to four characteristic bands belonging to the ring vibrations of a protonated pyridyl moiety are found between 1640 and 1460 cm−1.79,80 Typical for protonated pyridyl moieties, the band at ∼1640 cm−1 is the most pronounced one. The fingerprint regions of the IR spectra show the bands of the deformation vibrations of the methylene groups (∼1420–1390 cm−1), of the stretching vibrations of the PO3 moieties (∼1040–930 cm−1) and of the P–C stretching vibrations (∼790–750 cm−1).81 The latter seem to be insensitive to the state of deprotonation of the alkylphosphonic acid.82 The broad band near 900 cm−1 is very typical for solid acids and acid salts with very strong O–H⋯O hydrogen bonds which is sometimes named D type band.74 An example showing the identification of a D band spectrum is given by Bujak and Frank.83 A summary of tentative assignments of prominent IR bands is shown in the ESI† file.
Conclusions
Seven new salts in the 4,4′-bipyridine/alkane-α,ω-diphosphonic acid system have been prepared and characterised by single crystal X-ray diffraction and vibrational spectroscopy. In the case of [4,4′-H2bipy][H2O3P(CH2)6PO3H]2 (2), two concomitant polymorphs have been obtained and investigated with powder X-ray diffraction and differential scanning calorimetry.
Within the solids of the presented compounds, the 4,4′-bipyridine units were found to be either mono- or diprotonated. On the other hand, the counterions were hydrogen (ω-phosphonoalkyl)phosphonates in each case. Compound 7 might be considered as an addition compound of 4,4′-bipyridin-1-ium hydrogen (12-phosphonododecyl)phosphonate and the corresponding diphosphonic acid. Hence, for some of these compounds the principles of packing seem to outplay the stoichiometric conversion of the educts.
The common structural element of all [4,4′-Hbipy]+ salts presented in this contribution is an infinite hydrogen bonded chain of 4,4′-bipyridin-1-ium cations whereas the anionic substructures of these compounds are quite different. The crystal structures of the [4,4′-H2bipy][H2O3P(CH2)nPO3H]2 salts were classified into mainly two different types of structures. In both types, the stacking of anionic, hydrogen bonded strands is nearly identical. However, the π–π-stacked 4,4′-bipyridine-1,1′-diium cations can either lie in line with the anionic strands (type 1) or perpendicular to the aforementioned (type 2). The arrangement of type 2 results in a herringbone pattern. For [4,4′-H2bipy][H2O3P(CH2)6PO3H]2 (2), two polymorphs have been obtained which could be classified into type 1 and type 2, respectively. Only the structure of [4,4′-H2bipy][H2O3P(CH2)9PO3H]2 (4) could not be assigned to one of these two types.
Using the literature known plot of the valence of an O–H⋯O hydrogen bond against its O⋯O distance,84 the O⋯O distances within the range of 2.4 to 2.7 Å in the crystal structures of the compounds 1–7 must be attributed to valences of 0.50–0.25 for the H⋯O bond. Consequently, the O–H⋯O hydrogen bonds discussed in this contribution can be classified as strong to medium strong. For an evaluation of the strength of the N–H⋯O and N–H⋯N hydrogen bonds, a similar approach given for H⋯N distances can be used.84 This suggests that N⋯O and N⋯N distances close to 2.5 Å produce valences of more than 0.3 and thus should be associated with strong hydrogen bonds (for detailed geometric parameters, see the ESI† file).
Overall, hydrogen bonding appears as the front runner in the hierarchy of intermolecular interactions concerning the compounds discussed in this work followed by π–π-stacking and van der Waals forces. For example, the aforementioned bending of the anions' alkylene chains reveals that the hydrogen bonding between the –PO3H2 and –PO3H− groups outplays the van der Waals interactions within the hydrophobic regions. Nevertheless, the fact that a conformational disorder between the alkylene chains was not observed in any case indicates that van der Waals forces still provide a considerable contribution to the lattice enthalpy.
All the structures shown in this work, solely exhibit ‘pure’ stacks of anionic and cationic tectons. One of us (GJR) has already shown that α,ω-diammonioalkanes are feasible tectons for the creation of layered compounds including ‘mixed’ stacks of anions and cations.85 Hence, the formation of structures featuring ‘mixed’ stacks within the 4,4′-bipyridine/α,ω-diphosphonic acid system could possibly be achieved by using anionic tectons derived from diphosphonic acids with aromatic spacers like benzene-1,4-diphosphonic acid or biphenyl-4,4′-diphosphonic acid. Alternatively, hydrogen bonded networks with ‘mixed’ stacks can also be constructed from long-chained α,ω-diammonioalkanes and alkane-α,ω-diphosphonates as it has been suggested by Mahmoudkhani et al.35 The execution and examination of those intellectual pastimes is the next step of our continuing investigations within the fields of crystal engineering.
Acknowledgements
We thank E. Hammes and P. Roloff for technical support.
Notes and references
- S. Cheng and A. Clearfield, Appl. Catal., A, 1986, 26, 91 CrossRef CAS.
- C. Maillet, P. Janvier, M. Pipelier, T. Praveen, Y. Andres and B. Bujoli, Chem. Mater., 2001, 13, 2879 CrossRef CAS.
- O. R. Evans, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2001, 123, 10395 CrossRef CAS PubMed.
- H. L. Ngo, A. Hu and W. Lin, J. Mol. Catal. A: Chem., 2004, 215, 177 CrossRef CAS.
- B. Zhang and A. Clearfield, J. Am. Chem. Soc., 1997, 119, 2751 CrossRef CAS.
- J. Wu, H. Hou, H. Han and Y. Fan, Inorg. Chem., 2007, 46, 7960 CrossRef CAS PubMed.
- M. Plabst, L. B. McCusker and T. Bein, J. Am. Chem. Soc., 2009, 131, 18112 CrossRef CAS PubMed.
- D. Grohol, M. Subramanian, D. M. Poojary and A. Clearfield, Inorg. Chem., 1996, 35, 5264 CrossRef CAS.
- G. Alberti and M. Casciola, Solid State Ionics, 1997, 97, 177 CrossRef CAS.
- J. M. Taylor, R. K. Mah, I. L. Moudrakovski, C. I. Ratcliffe, R. Vaidhyanathan and G. K. Shimizu, J. Am. Chem. Soc., 2010, 132, 14055 CrossRef CAS PubMed.
- S. Kim, K. W. Dawson, B. S. Gelfand, J. M. Taylor and G. K. Shimizu, J. Am. Chem. Soc., 2013, 135, 963 CrossRef CAS PubMed.
- G. Cao and T. E. Mallouk, Inorg. Chem., 1991, 30, 1434 CrossRef CAS.
- A. Clearfield, C. K. Sharma and B. Zhang, Chem. Mater., 2001, 13, 3099 CrossRef CAS.
- S.-F. Tang, X.-B. Pan, X.-X. Lv, S.-H. Yan, X.-R. Xu, L.-J. Li and X.-B. Zhao, CrystEngComm, 2013, 15, 1860 RSC.
- M. Taddei, F. Costantino, A. Ienco, A. Comotti, P. V. Dau and S. M. Cohen, Chem. Commun., 2013, 49, 1315 RSC.
- D. Rong, H.-G. Hong, Y. I. Kim, J. S. Krueger, J. E. Mayer and T. E. Mallouk, Coord. Chem. Rev., 1990, 97, 237 CrossRef CAS.
- L. A. Vermeulen and M. E. Thompson, Chem. Mater., 1994, 6, 77 CrossRef CAS.
- H. Byrd, A. Clearfield, D. Poojary, K. P. Reis and M. E. Thompson, Chem. Mater., 1996, 8, 2239 CrossRef CAS.
- T.-Y. Ma, X.-J. Zhang, G.-S. Shao, J.-L. Cao and Z.-Y. Yuan, J. Phys. Chem. C, 2008, 112, 3090 CAS.
- H. Tian, Y.-Y. Zhu, Z.-G. Sun, F. Tong, J. Zhu, W. Chu, S.-H. Sun and M.-J. Zheng, New J. Chem., 2013, 37, 212 RSC.
- M.-J. Zheng, Y.-Y. Zhu, Z.-G. Sun, J. Zhu, C.-Q. Jiao, W. Chu, S.-H. Sun and H. Tian, CrystEngComm, 2013, 15, 1445 RSC.
- W. Chu, Z.-G. Sun, C.-Q. Jiao, Y.-Y. Zhu, S.-H. Sun, H. Tian and M.-J. Zheng, Dalton Trans., 2013, 42, 8009 RSC.
- G. Alberti, U. Costantino, S. Allulli and N. Tomassini, J. Inorg. Nucl. Chem., 1978, 40, 1113 CrossRef CAS.
- G. Cao, H. Lee, V. M. Lynch and T. E. Mallouk, Inorg. Chem., 1988, 27, 2781 CrossRef CAS.
- G. Alberti, R. Vivani and S. Murcia Mascarós, J. Mol. Struct., 1998, 470, 81 CrossRef CAS.
- H. P. Perry, J. Law, J. Zon and A. Clearfield, Microporous Mesoporous Mater., 2012, 149, 172 CrossRef CAS.
- P. O. Adelani and T. E. Albrecht-Schmitt, Cryst. Growth Des., 2011, 11, 4227 CAS.
- R.-B. Fu, X.-T. Wu, S.-M. Hu, J.-J. Zhang, Z.-Y. Fu and W.-X. Du, Inorg. Chem. Commun., 2003, 6, 827 CrossRef CAS.
- M. Taddei, F. Costantino, V. Manuali and R. Vivani, Inorg. Chem., 2011, 50, 10835 CrossRef CAS PubMed.
- M. J. Bialek, J. Janczak and J. Zon, CrystEngComm, 2013, 15, 390 RSC.
- J. Zon, D. Kong, K. Gagnon, H. Perry, L. Holliness and A. Clearfield, Dalton Trans., 2010, 39, 11008 RSC.
- K. D. Demadis, A. Panera, Z. Anagnostou, D. Varouhas, A. M. Kirillov and I. Císařová, Cryst. Growth Des., 2013, 13, 4480 CAS.
- M. Dines, R. Cooksey, P. Griffith and R. H. Lane, Inorg. Chem., 1983, 22, 1003 CrossRef CAS.
- C. K. Sharma, A. J. Hessheimer and A. Clearfield, Polyhedron, 2001, 20, 2095 CrossRef.
- A. H. Mahmoudkhani and V. Langer, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 2941 CrossRef CAS.
- A. H. Mahmoudkhani and V. Langer, J. Mol. Struct., 2002, 609, 97 CrossRef CAS.
- M. Plabst, N. Stock and T. Bein, Cryst. Growth Des., 2009, 9, 5049 CAS.
- A. H. Mahmoudkhani and V. Langer, Cryst. Growth Des., 2001, 2, 21 Search PubMed.
- J. K. Zaręba, M. J. Białek, J. Janczak, J. Zoń and A. Dobosz, Cryst. Growth Des., 2014, 14, 6143 Search PubMed.
- G. Ferguson, C. Glidewell, R. Gregson and P. Meehan, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 129 Search PubMed.
- R. M. Gregson, C. Glidewell, G. Ferguson and A. J. Lough, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 39 Search PubMed.
- P. S. Wheatley, A. J. Lough, G. Ferguson, C. J. Burchell and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 95 CAS.
- J. Fuller and N. E. Heimer, J. Chem. Crystallogr., 1995, 25, 129 CrossRef CAS.
- C. Glidewell, G. Ferguson and A. J. Lough, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 855 Search PubMed.
- W.-Q. Kan and J.-F. Ma, Z. Kristallogr. - New Cryst. Struct., 2011, 226, 73 CAS.
- C. K. Sharma and A. Clearfield, J. Am. Chem. Soc., 2000, 122, 4394 CrossRef CAS.
- W. Rui-Biao, F. Xin-Tao, H. Sheng-Min, D. Jian-Jun and Z. Wen-Xin, Chin. J. Struct. Chem., 2004, 23, 855 Search PubMed.
- J. Beckmann, R. Rüttinger and T. Schwich, Cryst. Growth Des., 2008, 8, 3271 CAS.
- M. K. Meyer, J. Graf and G. J. Reiß, Z. Naturforsch., B: J. Chem. Sci., 2010, 65, 1462 CAS.
- S. Peterson, E. Gebert, A. Reis Jr, M. Druyan, G. Mason and D. Peppard, J. Phys. Chem., 1977, 81, 466 CrossRef CAS.
- E. Gebert, A. Reis Jr, M. Druyan, S. Peterson, G. Mason and D. Peppard, J. Phys. Chem., 1977, 81, 471 CrossRef CAS.
- C. C. McLauchlan, A. J. Ziegler and A. W. Herlinger, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o1359 CAS.
- M. van Megen, W. Frank and G. J. Reiss, Z. Kristallogr. - Cryst. Mater., 2015, 230, 485 CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256 CrossRef.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- J. Grell, J. Bernstein and G. Tinhofer, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 1030 CrossRef.
- J. Grell, J. Bernstein and G. Tinhofer, Crystallogr. Rev., 2002, 8, 1 CrossRef CAS.
- G. Schwarzenbach and J. Zurc, Monatsh. Chem., 1950, 81, 202 CrossRef CAS.
- K. Moedritzer and R. Irani, J. Inorg. Nucl. Chem., 1961, 22, 297 CrossRef CAS.
- J. A. Griffith, D. J. McCauley, R. E. Barrans and A. W. Herlinger, Synth. Commun., 1998, 28, 4317 CrossRef CAS.
-
CrysAlisPro, 1.171.33.42, Oxford Diffraction Ltd., Oxford, U. K., 2009 Search PubMed.
-
W. Kraus and G. Nolze, PowderCell for Windows, 2.3, Berlin, Germany, 1999 Search PubMed.
-
Win-IR Pro, 2.97, Bio-Rad, Hercules, California, 1993 Search PubMed.
-
M. Feustel, Grundlagen der ATR-Technik, Resultec analytical equipment, Illerkirchberg, Germany, 1999 Search PubMed.
-
Opus, 6.5, Bruker, Billerica, Massachusetts, 2009 Search PubMed.
-
Callidus, 2E3, HEKAtech, Wegberg, Germany, 1998 Search PubMed.
-
STARe, 9.20, Mettler Toledo GmbH, Schwerzenbach, Switzerland, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- R. Boese, H.-C. Weiss and D. Bläser, Angew. Chem., Int. Ed., 1999, 38, 988 CrossRef CAS.
- G. J. Reiss and M. van Megen, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 5 CrossRef CAS.
- M. Bujak and W. Frank, Polyhedron, 2014, 68, 199 CrossRef CAS.
- D. Hadži, Pure Appl. Chem., 1965, 11, 435 CrossRef.
- M. F. Claydon and N. Sheppard, J. Chem. Soc. D, 1969, 1431 RSC.
- B. Engelen, K. Boldt, H. Müller and K. Unterweide, J. Mol. Struct., 1997, 436-437, 247 CrossRef CAS.
-
M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie, Thieme, Stuttgart, Germany, vol. 7, 2005 Search PubMed.
-
P. J. Larkin, IR and Raman Spectroscopy Principles and Spectral Interpretation, Elsevier, Amsterdam, Netherlands, vol. 1, 2011 Search PubMed.
- M. A. Martin-Drumel, O. Pirali, C. Falvo, P. Parneix, A. Gamboa, F. Calvo and P. Bréchignac, Phys. Chem. Chem.
Phys., 2014, 16, 22062 RSC.
- C. K. Pearce, D. W. Grosse and W. Hessel, J. Chem. Eng. Data, 1970, 15, 567 CrossRef CAS.
- L. van Haverbeke, H. O. Desseyn and M. A. Herman, Bull. Soc. Chim. Belg., 1972, 81, 547 CrossRef CAS.
- B. J. van der Veken and M. A. Herman, J. Mol. Struct., 1973, 15, 225 CrossRef CAS.
- M. Bujak and W. Frank, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 1391 CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- G. J. Reiss and J. S. Engel, CrystEngComm, 2002, 4, 155 RSC.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence