DOI:
10.1039/C5CE02109J
(Paper)
CrystEngComm, 2016,
18, 704-713
Isoskeletal Schiff base polynuclear coordination clusters: synthetic and theoretical aspects†
Received
28th October 2015
, Accepted 10th December 2015
First published on 17th December 2015
Abstract
This work addresses and enlightens synthetic aspects derived from our effort to systematically construct isoskeletal tetranuclear coordination clusters (CCs) of the general formula [TR2Ln2(LX)4(NO3)2(solv)2] possessing a specific defected dicubane topology, utilizing various substituted Schiff base organic ligands (H2LX) and NiII/CoII and Dy(OTf)3 salts. Our synthetic work is further supported by DFT studies.
Introduction
The chemistry of polynuclear coordination clusters (CCs)1,2 of paramagnetic 3d and/or 4f metals is an area of research that offers many interdisciplinary opportunities to connect with chemistry, physics, biology and materials science. Aesthetically pleasing structures have been reported;3–12 moreover, several research groups have showcased the relevance of CCs in a variety of fields such as catalysis,13–16 biology,17 luminescence,18–20 magnetic resonance21 and molecular magnetism.22,23
Several organic ligands have been employed to build new CCs24 and among them Schiff base ligands are widely employed to give access to a plethora of compounds with unprecedented topologies and interesting magnetic properties.5,8,9,25–45 The reason for the widespread selection of Schiff bases as ligands for the construction of polynuclear CCs is manifold: a) their facile synthesis and readily tunable structures obtainable in high yields; b) structural diversity is assured given the availability of a plethora of carbonyl and amine precursors; and c) the accessibility of several coordination sites allows for varied binding modes, although the exact structure and composition of the final product are invariably unpredictable. (E)-2-(2-Hydroxy-3-methoxybenzylideneamino)phenol H2L1 (Scheme 1, left) is derived from the condensation of o-vanillin and 2-aminophenol and several CCs bearing this ligand have been reported.46–57 The blending of this ligand with CoII or NiII and 4f nitrate salts results in tetranuclear CCs46,47 of the general formula [TR2Ln2L14(NO3)2(solv)2], where TR is CoII or NiII, Ln is DyIII or TbIII and solv is THF or DMF, possessing a defected dicubane or “butterfly”58 core (Scheme 1, middle) or a 2,3M4-1 (Scheme 1, right)48,59,60 topology with the 4f ions in the wings and the 3d metal in the hinge. This topology represents one of the most common structural motifs in magneto chemistry, and CCs of this type have been well-studied and their behaviour is often very well-understood.46,47 From a structural point of view, it is worth emphasizing that H2L1 supports this topology in the absence of any additional bridging atom.
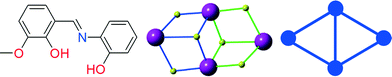 |
| | Scheme 1 (left) The (E)-2-(2-hydroxy-3-methoxybenzylideneamino)phenol H2L1 ligand, (middle) the defected dicubane motif, and (right) the 2,3M4-1 topology. | |
For a long time, synthetic chemists have employed new organic ligands along with 3d and/or 4f centres, under various reaction conditions, to afford new polynuclear species; however, few researchers have focused their efforts on tuning the periphery of the organic ligands and studying its influence on the properties of the final product. For example, Murugesu et al.35 reported a 7-fold enhancement of the energy barrier, Ueff, of a DyIII2 that behaves as Single Molecule Magnet (SMM), by introducing electron-withdrawing terminal ligands.
Having all this in mind, we initiated a project towards the synthesis of tetranuclear CCs of the general formula [TR2Ln2L14(NO3)2(solv)2], which possess the 2,3M4-1 topology, using CoII or NiII and 4f ions and substituted Schiff base ligands, similar to H2L1 (Scheme 2, left) in the presence or absence of substituted monocarboxylates as co-ligands (Scheme 2, right). The interest for this specific motif, which is supported only by the organic ligand in the absence of any additional bridging atom, is manifold: a) these types of molecules exhibit SMM behaviour46,47 and thus it is anticipated that the introduction of electron-withdrawing groups would improve their magnetic properties. b) Replacement of the 3d and 4f ions with luminescent elements such as ZnII and EuIII or TbIII, respectively, may lead to a new generation of materials with interesting optical properties. c) We recently showed that three tetranuclear CCs formulated as [TRII2DyIII2(L1)4(EtOH)6](ClO4)2·2(EtOH) where TR is CoII (1) or NiII (2) and [NiII2DyIII2(L1)4Cl2(CH3CN)2]·2(CH3CN) (3) display efficient homogeneous catalytic behaviour in the room-temperature synthesis of trans-4,5-diaminocyclopent-2-enones from 2-furaldehyde and primary or secondary amines under a non-inert atmosphere and thus, further tuning of the organic periphery may improve their catalytic performance.57 d) More importantly, the possibility of exchanging the 3d and/or the 4f elements with diamagnetic metal centres and retaining at the same time the structural topology in the solution gives the opportunity to study these molecules in the solid and/or solution state with several spectroscopic techniques such as NMR (1H, 13C, 89Y) or EPR for the Gd species.
 |
| | Scheme 2 (left) The substituted organic ligands used in this work R1 = H, Br, allyl, NO2; R2 = H, Cl, C6H5; R3 = H, NO2; R2−R3 = C4H4; (right) the substituted monocarboxylates used in this work (M1 R4 = R6=NO2; R5 = H; M2 R4 = NO2; R5 = R6 = H). | |
Adopting the isoreticular concept, introduced by O'Keeffe and Yaghi in metal–organic framework chemistry,61 a terminology that is not valid for finite structures, and since the isostructural and isomorphous terms do not fully describe the targeted CCs, we introduce herein the term “isoskeletal”, previously used to describe clusters that possess the same topology62,63 or related organic structures with the same host framework but different guests,64,65 to describe the targeted CCs constructed from organic ligands that provide a similar coordination environment and have the same 2,3M4-1 topology. This work addresses and enlightens synthetic and theoretical aspects derived from our effort to systematically construct isoskeletal CCs of the general formula [TR2Ln2(LX)4(NO3)2(solv)2], possessing the desired 2,3M4-1 topology. Magnetic, luminescence and catalytic studies of a few of the reported compounds will be discussed in future articles. To the best of our knowledge, such a systematic synthetic and theoretical investigation for the given NiII–CoII/DyIII species bearing the 2,3M4-1 topology has never been carried out.
Results and discussion
The synthesis of the substituted organic molecules represents the initial synthetic part of this project. A typical synthesis of a Schiff base ligand takes place in an alcoholic solution in the presence or absence of a catalyst (acid or base), although other synthetic methods such as microwave-mediated66 or solvent-free mechanochemical synthesis67,68 can be employed. Our attempts to synthesize the substituted organic molecules bearing electron-withdrawing groups (Scheme 2, left) (Table 1, entries 2–20) via a typical synthesis, viz. reflux in alcoholic solution of equivalent amounts of the corresponding aldehyde and amine, resulted in the desired products, but in very low yields. Microwave-assisted organic synthesis (MAOS) enables the rapid synthesis of organic molecules, often with excellent yields and selectivity. Interestingly, by applying MAOS69,70 for the synthesis of the corresponding organic molecules, the yield increases drastically. In total, we synthesized 19 new organic ligands (Table 1, entries 2–20) and we characterized via single crystal X-ray crystallography two examples, H2L3 and H2L19 (Fig. 1). Single crystal X-ray studies at 173 K reveal that both H2L3 and H2L19 are a mixture of keto–enol tautomers. In H2L3 (Fig. 1, above), where two molecules can be found in the asymmetric unit, the enol form of the ligand is indicative of the existence of protons H3 and H8A that could be freely refined with 22 and 23% chemical occupancy, respectively. However, to obtain a suitable structure solution, both protons were restrained to have 25% occupancy and their O–H bond distance to be at 0.88Å. The C–O bond distances are 1.282(3) and 1.282(3) Å which are slightly higher than a typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond. Similarly, for ligand H2L19, protons H1 and H2 (Fig. 1 below) could be freely refined and have 79% and 21% occupancies, indicating a 79/21 existence of the ketonic/enolic form, while the C–O bond distance is 1.285(2) Å. These findings are in line with previously reported compounds.67 Solution studies (1H and 13C NMR) of the synthesized compounds revealed the existence of both keto and enol forms.
O double bond. Similarly, for ligand H2L19, protons H1 and H2 (Fig. 1 below) could be freely refined and have 79% and 21% occupancies, indicating a 79/21 existence of the ketonic/enolic form, while the C–O bond distance is 1.285(2) Å. These findings are in line with previously reported compounds.67 Solution studies (1H and 13C NMR) of the synthesized compounds revealed the existence of both keto and enol forms.
Table 1 A list of all substituted Schiff bases synthesized for this study. R1, R2 and R3 correspond to the substituted groups seen in Scheme 2 (left)
| Entry |
|
R1 |
R2 |
R3 |
Yield (%) |
|
MW technique.
|
| 1 |
H2L1
|
H |
H |
H |
95 |
| 2 |
H2L2
|
H |
H |
NO2 |
66 [96]a |
| 3 |
H2L3
|
H |
Cl |
NO2 |
76 [95]a |
| 4 |
H2L4
|
H |
C6H5 |
H |
91 |
| 5 |
H2L5
|
H |
C4H4 |
|
88 |
| 6 |
H2L6
|
NO2 |
H |
H |
93 |
| 7 |
H2L7
|
NO2 |
H |
NO2 |
94 |
| 8 |
H2L8
|
NO2 |
Cl |
NO2 |
80 |
| 9 |
H2L9
|
NO2 |
C6H5 |
H |
92 |
| 10 |
H2L10
|
NO2 |
C4H4 |
|
74 |
| 11 |
H2L11
|
allyl |
H |
H |
70 [98]a |
| 12 |
H2L12
|
allyl |
H |
NO2 |
72 |
| 13 |
H2L13
|
allyl |
Cl |
NO2 |
70 |
| 14 |
H2L14
|
allyl |
C6H5 |
H |
69 |
| 15 |
H2L15
|
allyl |
C4H4 |
|
61 [95]a |
| 16 |
H2L16
|
Br |
H |
H |
84 |
| 17 |
H2L17
|
Br |
H |
NO2 |
79 |
| 18 |
H2L18
|
Br |
Cl |
NO2 |
35 [85]a |
| 19 |
H2L19
|
Br |
C6H5 |
H |
83 |
| 20 |
H2L20
|
Br |
C4H4 |
|
79 |
 |
| | Fig. 1 A schematic representation of H2L3 (up) and H2L19 (bottom) ligands. | |
The synthesis of the targeted tetranuclear CCs represents the next part of this project. Based on the synthetic protocols that resulted in the isolation of the targeted tetranuclear CCs [TR2Ln2(LX)4(NO3)2(solv)2],57 and bearing in mind that a subtle change in the synthetic procedure may affect the shape, dimensionality and nuclearity of the final product, we screened several molecular ratios to isolate the corresponding targeted isoskeletal CCs. At this point, it is worth mentioning that in order to avoid complicating the synthetic procedures we employed only Dy(OTf)3 as the lanthanide source. During this screening procedure several interesting and unexpected results were obtained, grouped and presented below.
A) Oxidation
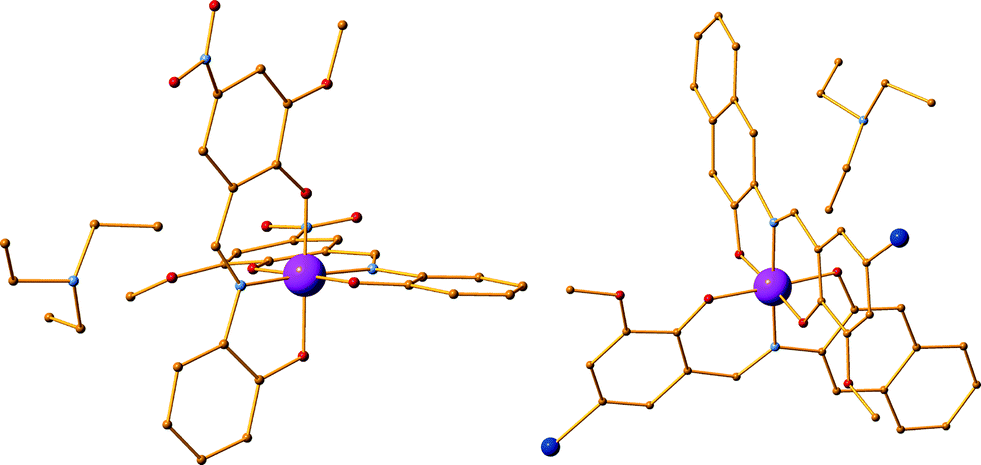
The reaction of H2L6 with Co(ClO4)2, Dy(OTf)3 and Et3N in the molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:5 in MeOH resulted, after 3 days, in approximately 82% yield, in dark-red shaped crystals formulated as (Et3NH)[CoIII(L6)2]·3MeOH [4·3MeOH] (Fig. 2 left). Similarly, the reaction of H2L20 with Co(ClO4)·6H2O, Dy(OTf)3 and Et3N in the molar ratio 1:1:1:3 in MeOH resulted, after 6 days, in approximately 82% yield dark, in red-shaped crystals formulated as (Et3NH)[CoIII(L20)2]·2MeOH [5·2MeOH] (Fig. 2, right). In the cases of 4 and 5, during crystallization, upon standing at room temperature, CoII is oxidized to CoIII. To target the isoskeletal 2,3M4-1 CoII analogues, it is important to perform the synthesis and crystallization under inert atmosphere conditions.
:1:1:5 in MeOH resulted, after 3 days, in approximately 82% yield, in dark-red shaped crystals formulated as (Et3NH)[CoIII(L6)2]·3MeOH [4·3MeOH] (Fig. 2 left). Similarly, the reaction of H2L20 with Co(ClO4)·6H2O, Dy(OTf)3 and Et3N in the molar ratio 1:1:1:3 in MeOH resulted, after 6 days, in approximately 82% yield dark, in red-shaped crystals formulated as (Et3NH)[CoIII(L20)2]·2MeOH [5·2MeOH] (Fig. 2, right). In the cases of 4 and 5, during crystallization, upon standing at room temperature, CoII is oxidized to CoIII. To target the isoskeletal 2,3M4-1 CoII analogues, it is important to perform the synthesis and crystallization under inert atmosphere conditions.
 |
| | Fig. 2 (left) The crystal structure of compound 4. (right) The crystal structure of compound 5. Solvent molecules and H-atoms are omitted for clarity. Colour code: Co (pink), N (light blue), O (red), C (orange), Br (blue). | |
B) Solvent influence
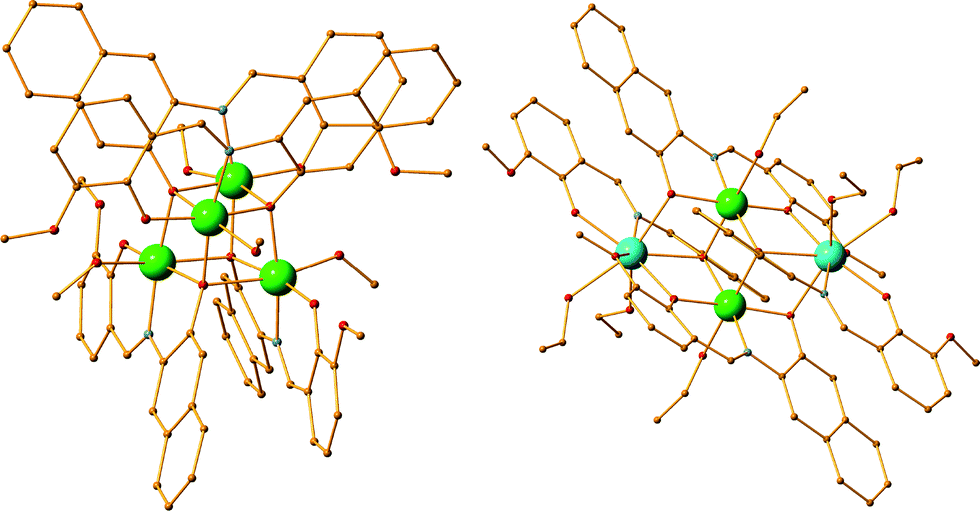
As anticipated, modification of the organic periphery of the parent organic ligand H2L1 drastically affects its solubility. However, when targeting the corresponding isoskeletal tetranuclear species, the use of a mixture of solvents, to enhance dissolution, results in the formation of undesired compounds. For example, the reaction of ligand H2L14 with Ni(ClO4)2, Dy(OTf)3 and Et3N in the molar ratio 2:1:1:5 in a mixture of EtOH/CH2Cl2 results in the synthesis of a tetranuclear compound formulated as [NiII2DyIII2(L14)4(allyl-o-vanillin)2(EtOH)2]·3CH2Cl2 [6·3CH2Cl2] (Fig. 3). The two allyl-o-vanillin moieties are derived from the disassembly of H2L14 during the reaction. Interestingly, performing a similar reaction in solely DMF results in the formation of the desired [NiII2DyIII2(L14)4(DMF)6]·2(ClO4)·4DMF [7·4DMF] (Fig. 3). This observation was additionally proven by performing a reaction of ligand H2L19 with Ni(ClO4)2·6H2O, Dy(OTf)3 and Et3N in the molar ratio 2:1:1:5 in a mixture of EtOH/CH2Cl2, which resulted in the isolation of compound [NiII2DyIII2(L19)4(bromo-o-vanillin)2(EtOH)2]·6EtOH [8·6EtOH] (Fig. 3). Moreover, utilizing a blend of EtOH/THF in the following reaction of Ni(ClO4)2·6H2O, Dy(OTf)3 and Et3N in the molar ratio 2:1:1:5 results in [NiII2DyIII2(L15)4(allyl-o-vanillin)2(EtOH)2]·2THF·2EtOH [9·2THF·2EtOH] (Fig. 3). However, performing a similar reaction in DMF results in the formation of the desired compound [NiII2DyIII2(L15)4(DMF)2]·2(ClO4) [10] (Fig. 3). The aforementioned examples indicate that the use of a second solvent increases the chances of the formation of a by-product and thus the mixture of solvents may not correspond to an ideal synthetic protocol when targeting high purity compounds.
 |
| | Fig. 3 The crystal structures of compounds 6–10. H atoms and solvents are omitted for clarity. The substituted o-vanillin moiety is indicated with blue colour. Colour code: Ni (light green), Dy (light blue), N (blue), O (red), C (orange), Br (blue). | |
C) Solvent concentration influence
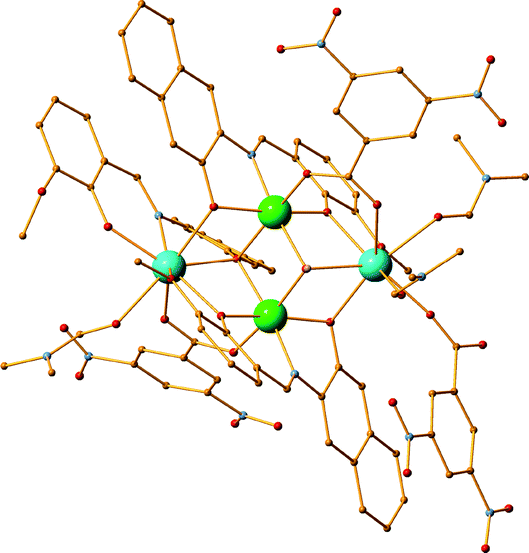
During our efforts to scale up the synthesis of compound 2,57 we performed reactions with a lower amount of solvent (10 ml instead of 20 ml) or we tripled the amount of the reactants while retaining the same the molar ratio, but then a different product was obtained, formulated as [NiII2DyIII2(L1)4(EtOH)4(H2O)2](ClO4)2 (11) (Fig. 4). Two water molecules coordinate to NiII centres in 11, in contrast to two ethanol molecules found in 2.57 Moreover, when a similar scale up reaction is performed in a different solvent such as DMF, another compound is formed formulated as [NiII2DyIII2(L1)4(DMF)6](OTf3)2·2DMF (12·2DMF) (Fig. 4), instead of the expected perchlorate derivative. In addition, performing a concentrated reaction of Ni(ClO4)·6H2O, Dy(OTf)3, Et3N and H2L20 in the molar ratio 1:1:2:8 resulted in a CC with the formula [NiII2DyIII2(L20)4(DMF)6] [NiII2DyIII2(L20)4(DMF)4(H2O)2]·4(ClO4)·5DMF (13·5DMF) (Fig. 4).
 |
| | Fig. 4 The crystal structures of 11, 12 and 13. Colour code: Ni (light green), Dy (light blue), N (blue), O (red), C (orange), Br (blue), F (green) S (yellow). | |
D) Molar ratio
The reaction of Ni(ClO4)2·6(H2O), Dy(OTf)3, and H2L5 with Et3N in MeOH in the molar ratio 1:1:2:5 resulted in the isolation of a tetranuclear cubane Ni4 formulated as [NiII4(L5)4(MeOH)4]·6MeOH (14·6MeOH) which is isoskeletal to the tetranuclear cubic Ni4 CC formed by the H2L1 ligand, reported recently.54 However, performing the same reaction in EtOH results in the isolation of the targeted isoskeletal tetranuclear [NiII2DyIII2(L5)4(EtOH)6]·2(ClO4)·4EtOH (15·4EtOH) (Fig. 5).
 |
| | Fig. 5 The crystal structures of compounds 14 (left) and 15 (right). Colour code: Ni (light green), Dy (light blue), N (blue), O (red), C (orange). | |
E) Solvent diffusion crystallization
In our effort to obtain the targeted tetranuclear NiII2DyIII2 analogues with H2L4 as a ligand via the diffusion crystallization technique, a compound formulated as [NiII2DyIII2(L4)4(DMF)6]·2(ClO4)·2Et2O (16·2Et2O) (Fig. 6, left) which contains two ether molecules as lattice solvents, was isolated. Upon standing at room temperature,71–74 compound 16 immediately loses its crystallinity.
 |
| | Fig. 6 The crystal structures of compound 16 (left) and 17 (right). Colour code: Ni (light green), Dy (light blue), N (blue), O (red), Cl (light green). | |
F) Co-ligand introduction
To identify the possibility of introducing a monocarboxylate group as a co-ligand on the periphery of the 2,3M4-1 motif, we performed a pilot reaction with H2L1, Ni(OAc)2·4(H2O), Dy(OTf)3 and Et3N in the molar ratio 2:1:1:2 in MeOH that resulted in the isolation of a compound formulated as [NiII2DyIII2(L1)4(OAc)2(MeOH)2]·2MeOH (17·2MeOH) (Fig. 6, right). Each acetate group bridges the Ni and Dy centres, which is in contrast to the NO3 analogues that prefer to chelate to the Ln centres.46,47
Employment of the bulky co-ligands M1 and M2 along with ligand H2L12, Co(ClO4)2·6H2O and Dy(OTf)3 in the molar ratio of 2:1:1:5 in N,N′-DMF as solvent results in the isolation of two compounds formulated as [CoII2DyIII2(L12)4(M1)2(DMF)2] (18) and [CoII2DyIII2(L12)4(M2)2(DMF)2]·2DMF (19·2DMF), respectively (Fig. 7).
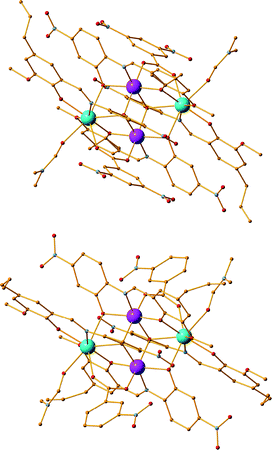 |
| | Fig. 7 The crystal structures of compounds 18 (upper) and 19 (lower). Colour code: Co (pink), Dy (light blue), N (blue), O (red), C (orange). | |
However, our effort to obtain the isoskeletal compound to 18 CoII2DyIII2 compound along with ligand H2L2 instead of H2L12 we performed a similar reaction which resulted in red plate crystals formulated as [DyIII(M1)3(DMF)2] (20) (Fig. 8). Compound 20 is a one-dimensional (1D) coordination polymer and can be considered as a polymorph to the recently reported compound.75
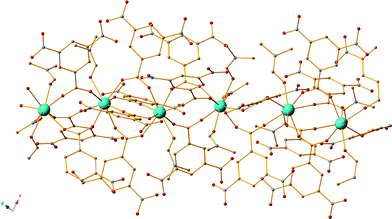 |
| | Fig. 8 The crystal structure of compound 20. Colour code: Dy (light blue), N (blue), O (red), C (orange). | |
Finally, the diffusion of Et2O to a concentrated reaction of ligand H2L5 along with Ni(ClO4)·6H2O, Dy(OTf)3, M1 and Et3N in DMF results in the formation of a compound formulated as [NiII2DyIII2(OH)(L5)3(M1)3(DMF)2]·1.5DMF·Et2O (21·1.5DMF·Et2O) which possesses the 2,3M4-1 topology. Surprisingly, only three organic ligands are involved in the aggregation in contrast to the four ligands used in compounds 6–19. The skeleton of 21 is further supported by the presence of a hydroxyl μ3-OH group. Also, one Et2O molecule is trapped in the lattice (Fig. 9).
 |
| | Fig. 9 The crystal structure of compound 21. Colour code: Ni (light green), Dy (light blue), N (blue), O (red), C (orange), H (white). | |
Solution studies
To further confirm the identity of compounds 7, 10 and 16 in solution, we performed an electrospray ionization mass (ESI-MS) spectrometry study. For 7, we observed three peaks in the MS (positive-ion mode) at m/z 1009.2113, 972.1730 and 936.1485 which correspond perfectly to three dicationic fragments, [NiII2DyIII2(L14)4(DMF)2]2+, [NiII2DyIII2(L14)4(DMF)]2+ and [NiII2DyIII2(L14)4]2+, respectively (see Fig. S1 ESI†). Similarly, for 10, the peaks at m/z 1742.2549, 876.6074, 840.0702 and 803.067 correspond to [NiII2DyIII2(L15)4(MeOH)]+, [NiII2DyIII2(L15)4(DMF)2]2+, [NiII2DyIII2(L15)4(DMF)]2+, and [NiII2DyIII2(L15)4]2+ fragments (see Fig. S2†), respectively. For compound 16, the following four m/z 1847.228, 929.1356, 892.1121 and 855.5781 peaks correspond to [NiII2DyIII2(L4)4(MeOH)]+, [NiII2DyIII2(L4)4(DMF)2]2+, [NiII2DyIII2(L4)4(DMF)]2+ and [NiII2DyIII2(L4)4]2+ fragments (see Fig. S3†), respectively. However, all the efforts to obtain similar ESI-MS spectra for compounds 18 and 19, which contain a co-ligand, were not successful.
Theoretical studies
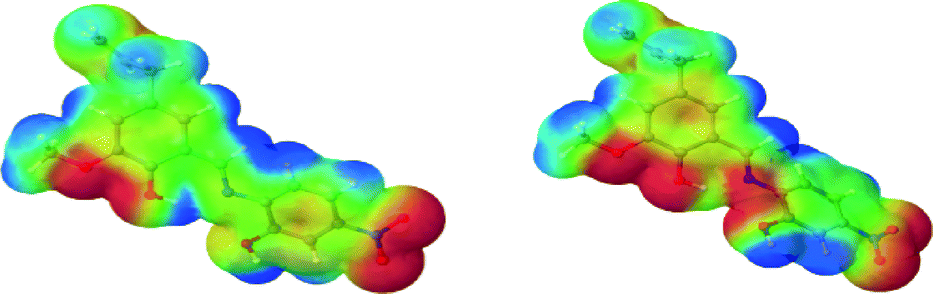
To gain further insight into the tuning and modulation of, inter alia, the catalytic activities of the tetranuclear CCs, computational studies were carried out. Specifically, the influence of the organic ligands on the electronic properties of the metals was studied. The reason for this study is that the ‘activity’ of the ligands may be tuned by employing diverse functional groups at specific sites/positions. In order to do this, quantitative and qualitative metrics of the functional group effects were established and in parallel several congeners of the ligands were examined; more so, several model systems of the polynuclear coordination clusters were used. With respect to Scheme 2 (see text) concerning the numbering, the following tables (Tables 2–5) indicate the model systems studied. Fig. 10 shows the visualization of the mapped molecular electrostatic potential in ligands L1m to L12m, as in the usual molecular electrostatic potential (MEP) rendering, with the blue zones depicting the ‘positively’ charged zones, while the red zones the ‘negatively charged zones’.
Table 2 Recapitulative table of the model ligands used
| L |
R1 |
R2 |
R3 |
| L1m |
H |
H |
H |
| L2m |
OMe |
H |
H |
| L3m |
N(CH2CH3)2 |
H |
H |
| L4m |
F |
H |
H |
| L5m |
H |
H |
NO2 |
| L6m |
CH2CHCH2 |
H |
NO2 |
| L7m1 |
NO2 |
H |
H |
| L8m |
NO2 |
H |
H |
| L9m |
H |
Cl |
NO2 |
| L10m |
Br |
H |
NO2 |
| L11m |
NO2 |
H |
NO2 |
| L12m |
NO2 |
Cl |
NO2 |
| L7: an additional NO2 is substituted at the ortho position adjacent to R2. |
Table 3 Recapitulative table of the co-ligands used
| coL |
R4 |
R5 |
R6 |
| coL1m |
H |
H |
H |
| coL2m |
H |
F |
H |
| coL3m |
NO2 |
H |
NO2 |
Table 4 Recapitulative table of the models used
| Model |
M1 |
M2 |
L |
coL |
S |
| m1 11 |
Zn |
Y |
L1m |
coL2m |
(CH3)2NCOH |
| m2 11a |
Ni |
Y |
L1m |
coL2m |
(CH3)2NCOH |
| m3 12 |
Zn |
Y |
L5m |
MeOH |
NO3 |
| m4 12a |
Ni |
Y |
L5m |
MeOH |
NO3 |
| m5 13 |
Zn |
Y |
L1m |
EtOH1 |
— |
| m6 14 |
Zn |
Y |
L6m |
coL3m |
S1m |
| m7 14b |
Zn |
Y |
L1m |
coL3m |
S1m |
Table 5 Mulliken charges
| Model |
q(M1) |
q(M2) |
q(O1) |
q(N) |
q(O2) |
| m1 11 |
0.79 |
1.49 |
−0.64 |
−0.20 |
−0.61 |
| m2 11a |
0.53 |
1.49 |
−0.61 |
−0.16 |
−0.59 |
| m3 12 |
0.93 |
1.34 |
−0.55 |
−0.26 |
−0.73 |
| m4 12a |
0.69 |
1.36 |
−0.62 |
−0.17 |
−0.58 |
| m5 13 |
0.86 |
1.32 |
−0.56 |
−0.30 |
−0.73 |
| m6 14 |
0.85 |
1.56 |
−0.55 |
−0.27 |
−0.70 |
| m7 14b |
0.85 |
1.55 |
−0.56 |
−0.27 |
−0.71 |
 |
| | Fig. 10 Rendition of the molecular electrostatic potential (MEP) of the model ligands. | |
One way to assess the effect of the electron-withdrawing groups on the Schiff base is to qualitatively render the MEP mapped onto the van der Waals surface. Indeed, the nature of the R groups has a profound effect on the surface charge of the base on its ‘inner cove’ – that is, the O–O–N–O core pattern that directly interacts with the metals. Fig. 10 and Table 5 qualitatively illustrate this idea. Unsurprisingly, the results show that the charge distribution along the cove can indeed be modulated upon modification of the groups.
In the gas phase, in their most stable geometrical configuration, the most ‘simple’ functionalization, i.e. having R1 = R2 = R3 = H turns out to confer the highest negative charge accumulation on the ligand ‘anchor’ points, the least being with R1 = NO2, R2 = Cl, R3 = NO2. Interestingly, for a given R1, changing R3 has a profound effect on the charge distribution as can be illustrated by considering L1m and L5m. On the other hand, for a given R3, changing R1 does not have a noticeable altering effect, as shown for example by L5m versus L10m and L1m versus L2m. More crucially, the conformation of the ligand, although not surprisingly, ultimately determines the extent, nature and motifs of the interaction between the metal-interacting side of the ligands and the metals (see Fig. 11). Indeed, single point (sp) calculations on L6m in its ligand-bound geometry exhibits a different charge distribution signature with respect to that of its gas-phase optimized structure (gp). The geometries differ by having the nitrogen-bound aryl group (ring 2) rotated clockwise (gp) or counter clockwise (sp) around the N–C(aryl) bond. This observation can be rationalized when one considers the frontier molecular orbitals (MO).
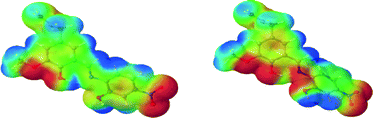 |
| | Fig. 11 Rendition of the molecular electrostatic potential (MEP) of L6m in its ligand-bound (left) and in its free conformation (right). | |
Indeed, inspection of the LUMO–HOMO-2 of L6m (sp) indicates that the molecular orbitals (MO) are mainly consisted of π networks running throughout the whole molecule; even more remarkable is the low mixing of atomic p orbitals perpendicular to the molecular plane defined by ring 1 (the aryl containing R1) and the N-centered p orbital that is parallel to this plane (see Fig. 12). This very orientation of the p orbital should also be responsible for the dependence of the cove charge distribution on the orientation of the hydrogens on the OH functions on both rings (data not shown).
 |
| | Fig. 12 Frontier orbitals of L6m (sp). | |
In terms of partial atomic charge, within the Mulliken partitioning scheme, it can be noted that there are indeed variations in values on the metals, on the two oxygen and the nitrogen atoms (see Table 5) depending on the makeup of the ligand. Close examination suggests that, in terms of partial charges, both M2 (Table 4) and O1 (the phenolic oxygen atom of the o-vanillin moiety) are spectator elements and the value of their partial charge does not depend on that of the other atoms. However, an interdependence of the values of partial charge on N, O2 (the phenolic oxygen atom of the aminophenol moiety) and M1 can be found. The largest of such variations occurs for metal M1 and is observed between model m2 11a and model m3 12. These results are perfectly in line with that of the charge distribution being dependent on the orientation of ring 2.
The above results clearly show the richness of the chemistry of the present coordination clusters: the nature of metal M1 and the identity of the R groups in the ligands play preponderant roles in both the tuning and control of the behaviour of the clusters. With the trends and rationale hereby demonstrated, it should be possible in future work to better modulate the activity of the clusters through tuning of the electronic structure and control of structural constraints.
Conclusions
In this work we present the synthesis of nineteen new substituted Schiff base organic ligands and eighteen new CCs. Adopting the MOF isoreticular concept, we introduce herein the term “isoskeletal” which ideally describes the structural and topological similarities found in compounds 6–13, 15–19 and 21. Microwave synthesis has proven to be very efficient for the synthesis of the substituted Schiff base ligands.76 Targeting the synthesis of such isoskeletal species, it is shown that several parameters must be taken into account. We do show here that a slight change in the synthetic procedure drastically affects the formula of the desired compound. However, the usage of different lanthanide sources and the lanthanide contraction are two parameters that have not been taken into account in this study and are anticipated to have a major impact on the shape, nuclearity and formula of the final product. The possibility of producing isoskeletal species and exchanging the 3d and/or the 4f elements with diamagnetic elements such as ZnII or YIII gives the opportunity to further study the targeted molecules in the solid and/or solution state with additional spectroscopic techniques such as NMR (1H, 13C, 89Y) or EPR for the GdIII species. For the present study, ESI-MS studies of compounds 7, 10 and 16 are indicative that the targeted species remain intact in solution. However, upon introduction of co-ligands the targeted CCs become insoluble in alcoholic solvents. Computational studies showcase how the likely activity of the CCs can be tuned and modulated, and suggest ways on achieving this, hence demonstrating the richness of the chemistry of the hereby reported constructs. Our future efforts will be focused on further exploring the coordination chemistry of these species as well on performing such systematic studies involving other organic ligands, aiming to isolate molecules with better magnetic, catalytic or fluorescence properties.
Experimental
Materials
Chemicals (reagent grade) were purchased from Sigma Aldrich and Alfa Aesar. Detailed synthesis and characterization of the HL ligand is described in the ESI.† All experiments were performed under aerobic conditions using materials and solvents as received. Safety note: perchlorate salts are potentially explosive; such compounds should be used in small quantities and handled with caution and utmost care at all times.
Synthesis
Detailed synthetic protocols can be found in the ESI.† Powder XRD spectra of compounds 4–21 were not recorded, due to the fact that these molecules can be considered as unexpected reaction products. Elemental analysis results of selected samples (7, 10, 11, 13 and 16) are reported in the ESI.†
Instrumentation
IR spectra of the samples were recorded over the range of 4000–650 cm−1 on a Perkin Elmer Spectrum One FT-IR spectrometer fitted with a UATR polarization accessory.
Computational details
Energy minimization on strategically designed model compounds (see Results and discussion) was conducted within the Kohn–Sham Density Functional Theory (DFT) approach at the B3LYP/SDD and B3LYP/6-311G* levels.77–87 Calculations were carried using the Gaussian09 (ref. 88) software. The Jmol program was used for visualization purposes.89
X-ray crystallography
Data for H2L3, H2L19, 5–8, 10–12, 15–17 and 21 were collected (ω-scans) at the University of Sussex using an Agilent Xcalibur Eos Gemini Ultra diffractometer with a CCD plate detector under a flow of nitrogen gas at 173(2) K using Mo Kα radiation (λ = 0.71073 Å). CRYSALIS CCD and RED software were used respectively for data collection and processing. Reflection intensities were corrected for absorption by the multi-scan method. Data for 4, 9, 13, 14, 18–20 were collected at the National Crystallography Service, University of Southampton.90 All structures were determined using Olex2,91 solved using either Superflip92 or SHELXT93,94 and refined with SHELXL-2014.95 All non-H atoms were refined with anisotropic thermal parameters, and H-atoms were introduced at calculated positions and allowed to ride on their carrier atoms. Geometric/crystallographic calculations were performed using PLATON,96 Olex2,91 and WINGX93 packages; graphics were prepared with Crystal Maker.97 For compounds 8, 9 and 14 the SQUEEZE process was applied.98 Each of the crystal structures has been deposited at the CCDC with reference numbers 1406218–1406237.
Acknowledgements
We thank the EPSRC (UK) for funding (grant number EP/M023834/1), the University of Sussex for offering a PhD position to K. G., the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for compounds 4, 9, 13, 14, 18–20, the Research Development Funding from the University of Sussex (V. N. D.) and Profs. Vladislav Blatov and Davide Proserpio for helpful scientific discussions.
Notes and references
-
L. Cronin and J. Fielden, in Coordination Clusters in Encyclopedia of Supramolecular Chemistry, Taylor and Francis, London, 2007, Taylor & Francis, 2007, pp. 1–10 Search PubMed.
- G. E. Kostakis, A. M. Ako and A. K. Powell, Chem. Soc. Rev., 2010, 39, 2238–2271 RSC.
- Z.-M. Zhang, S. Yao, Y.-G. Li, R. Clérac, Y. Lu, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2009, 131, 14600–14601 CrossRef CAS PubMed.
- X.-J. Kong, Y.-P. Ren, W.-X. Chen, L.-S. Long, Z. Zheng, R.-B. Huang and L.-S. Zheng, Angew. Chem., Int. Ed., 2008, 47, 2398–2401 CrossRef CAS PubMed.
- A. A. Athanasopoulou, M. Pilkington, C. P. Raptopoulou, A. Escuer and T. C. Stamatatos, Chem. Commun., 2014, 50, 14942–14945 RSC.
- C.-M. Liu, D.-Q. Zhang and D.-B. Zhu, Dalton Trans., 2010, 39, 11325–11328 RSC.
- J.-B. Peng, X.-J. Kong, Q.-C. Zhang, M. Orendáč, J. Prokleška, Y.-P. Ren, L.-S. Long, Z. Zheng and L.-S. Zheng, J. Am. Chem. Soc., 2014, 136, 17938–17941 CrossRef PubMed.
- Z. M. Zhang, L. Y. Pan, W. Q. Lin, J. D. Leng, F. S. Guo, Y. C. Chen, J. L. Liu and M. L. Tong, Chem. Commun., 2013, 49, 8081–8083 RSC.
- L. Zhao, J. Wu, S. Xue and J. Tang, Chem. - Asian J., 2012, 7, 2419–2423 CrossRef CAS PubMed.
- M. Wu, F. Jiang, D. Yuan, J. Pang, J. Qian, S. A. Al-Thabaiti and M. Hong, Chem. Commun., 2014, 50, 1113–1115 RSC.
- D. D'Alessio, A. N. Sobolev, B. W. Skelton, R. O. Fuller, R. C. Woodward, N. A. Lengkeek, B. H. Fraser, M. Massi and M. I. Ogden, J. Am. Chem. Soc., 2014, 136, 15122–15125 CrossRef PubMed.
- E. N. Chygorin, V. N. Kokozay, I. V. Omelchenko, O. V. Shishkin, J. Titiš, R. Boča and D. S. Nesterov, Dalton Trans., 2015, 44, 10918–10922 RSC.
- D. S. Nesterov, E. N. Chygorin, V. N. Kokozay, V. V. Bon, R. Boča, Y. N. Kozlov, L. S. Shul'pina, J. Jezierska, A. Ozarowski, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2012, 51, 9110–9122 CrossRef CAS PubMed.
- Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 4048–4058 RSC.
- G. Maayan and G. Christou, Inorg. Chem., 2011, 50, 7015–7021 CrossRef CAS PubMed.
- D. S. Nesterov, V. N. Kokozay, J. Jezierska, O. V. Pavlyuk, R. Boča and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 4401–4411 CrossRef CAS PubMed.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- D. I. Alexandropoulos, L. Cunha-Silva, L. Pham, V. Bekiari, G. Christou and T. C. Stamatatos, Inorg. Chem., 2014, 53, 3220–3229 CrossRef CAS PubMed.
- X. Yang, Z. Li, S. Wang, S. Huang, D. Schipper and R. A. Jones, Chem. Commun., 2014, 50, 15569–15572 RSC.
- J. Jankolovits, C. M. Andolina, J. W. Kampf, K. N. Raymond and V. L. Pecoraro, Angew. Chem., Int. Ed., 2011, 50, 9660–9664 CrossRef CAS PubMed.
- G. Guthausen, J. R. Machado, B. Luy, A. Baniodeh, A. K. Powell, S. Krämer, F. Ranzinger, M. P. Herrling, S. Lackner and H. Horn, Dalton Trans., 2015, 44, 5032–5040 RSC.
- E. C. Sañudo and L. Rosado Piquer, Dalton Trans., 2015, 44, 8771–8780 RSC.
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74–122 CrossRef CAS.
- A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC.
- L. Zhao, J. Wu, H. Ke and J. Tang, Inorg. Chem., 2014, 53, 3519–3525 CrossRef CAS PubMed.
- V. Chandrasekhar, P. Bag, W. Kroener, K. Gieb and P. Müller, Inorg. Chem., 2013, 52, 13078–13086 CrossRef CAS PubMed.
- S. Hossain, S. Das, A. Chakraborty, F. Lloret, J. Cano, E. Pardo and V. Chandrasekhar, Dalton Trans., 2014, 43, 10164–10174 RSC.
- V. Chandrasekhar, S. Das, A. Dey, S. Hossain, F. Lloret and E. Pardo, Eur. J. Inorg. Chem., 2013, 2013, 4506–4514 CrossRef CAS.
- M. Nematirad, W. J. Gee, S. K. Langley, N. F. Chilton, B. Moubaraki, K. S. Murray and S. R. Batten, Dalton Trans., 2012, 41, 13711–13715 RSC.
- M. Towatari, K. Nishi, T. Fujinami, N. Matsumoto, Y. Sunatsuki, M. Kojima, N. Mochida, T. Ishida, N. Re and J. Mrozinski, Inorg. Chem., 2013, 52, 6160–6178 CrossRef CAS PubMed.
- M. Sarwar, A. M. Madalan, C. Tiseanu, G. Novitchi, C. Maxim, G. Marinescu, D. Luneau and M. Andruh, New J. Chem., 2013, 37, 2280–2292 RSC.
- L.-L. Fan, F.-S. Guo, L. Yun, Z.-J. Lin, R. Herchel, J.-D. Leng, Y.-C. Ou and M.-L. Tong, Dalton Trans., 2010, 39, 1771–1780 RSC.
- L. Zhao, S. Xue and J. Tang, Inorg. Chem., 2012, 51, 5994–5996 CrossRef CAS PubMed.
- L.-F. Zou, L. Zhao, Y.-N. Guo, G.-M. Yu, Y. Guo, J. Tang and Y.-H. Li, Chem. Commun., 2011, 47, 8659–8661 RSC.
- F. Habib, G. Brunet, V. Vieru, I. Korobkov, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 13242–13245 CrossRef CAS PubMed.
- J.-P. Costes and C. Duhayon, Eur. J. Inorg. Chem., 2014, 4745–4749 CrossRef CAS.
- V. Gómez, L. Vendier, M. Corbella and J.-P. Costes, Inorg. Chem., 2012, 51, 6396–6404 CrossRef PubMed.
- E. Loukopoulos, B. Berkoff, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, A. Escuer and G. E. Kostakis, Eur. J. Inorg. Chem., 2015, 2646–2649 CrossRef CAS.
- B. Berkoff, K. Griffiths, A. Abdul-Sada, G. J. Tizzard, S. Coles, A. Escuer and G. E. Kostakis, Dalton Trans., 2015, 44, 12788–12795 RSC.
- M. Andruh, Dalton Trans., 2015, 44, 16633–16653 RSC.
- J. Wu, L. Zhao, P. Zhang, L. Zhang, M. Guo and J. Tang, Dalton Trans., 2015, 44, 11935–11942 RSC.
- H. Wang, H. Ke, S.-Y. Lin, Y. Guo, L. Zhao, J. Tang and Y.-H. Li, Dalton Trans., 2013, 42, 5298–5303 RSC.
- P. Zhang, L. Zhang, S.-Y. Lin and J. Tang, Inorg. Chem., 2013, 52, 6595–6602 CrossRef CAS.
- H. Ke, L. Zhao, Y. Guo and J. Tang, Dalton Trans., 2012, 41, 2314–2319 RSC.
- H. Ke, L. Zhao, Y. Guo and J. Tang, Dalton Trans., 2012, 41, 9760–9765 RSC.
- K. C. Mondal, A. Sundt, Y. Lan, G. E. Kostakis, O. Waldmann, L. Ungur, L. F. Chibotaru, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2012, 51, 7550–7554 CrossRef CAS PubMed.
- K. C. Mondal, G. E. Kostakis, Y. Lan, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2011, 50, 11604–11611 CrossRef CAS PubMed.
- K. C. Mondal, G. E. Kostakis, Y. Lan and A. K. Powell, Polyhedron, 2013, 66, 268–273 CrossRef CAS.
- H. Ke, L. Zhao, Y. Guo and J. Tang, Inorg. Chem., 2012, 51, 2699–2705 CrossRef CAS PubMed.
- H. Ke, W. Zhu, S. Zhang, G. Xie and S. Chen, Polyhedron, 2015, 87, 109–116 CrossRef CAS.
- R. Kannappan, D. M. Tooke, A. L. Spek and J. Reedijk, Inorg. Chim. Acta, 2006, 359, 334–338 CrossRef CAS.
- I. Nemec, M. Machata, R. Herchel, R. Boča and Z. Trávníček, Dalton Trans., 2012, 41, 14603–14610 RSC.
- N. Kushwah, M. K. Pal, A. Wadawale, V. Sudarsan, D. Manna, T. K. Ghanty and V. K. Jain, Organometallics, 2012, 31, 3836–3843 CrossRef CAS.
- S. Saha, S. Pal, C. J. Gómez-García, J. M. Clemente-Juan, K. Harms and H. P. Nayek, Polyhedron, 2014, 74, 1–5 CrossRef CAS.
- M. Hasanzadeh, M. Salehi, M. Kubicki, S. M. Shahcheragh, G. Dutkiewicz, M. Pyziak and A. Khaleghian, Transition Met. Chem., 2014, 39, 623–632 CrossRef CAS.
- D. Dey, G. Kaur, A. Ranjani, L. Gayathri, P. Chakraborty, J. Adhikary, J. Pasan, D. Dhanasekaran, A. R. Choudhury, M. A. Akbarsha, N. Kole and B. Biswas, Eur. J. Inorg. Chem., 2014, 3350–3358 CrossRef CAS.
- K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed.
- P. King, R. Clerac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Dalton Trans., 2004, 2670–2676 RSC.
- G. E. Kostakis and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2686–2697 CrossRef CAS.
- G. E. Kostakis, V. A. Blatov and D. M. Proserpio, Dalton Trans., 2012, 41, 4634–4640 RSC.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- R. Della Pergola, F. Demartin, L. Garlaschelli, M. Manassero, S. Martinengo, N. Masciocchi and D. Strumolo, Inorg. Chem., 1991, 30, 846–849 CrossRef CAS.
- D. Collini, F. F. De Biani, S. Fedi, C. Femoni, F. Kaswalder, M. C. Iapalucci, G. Longoni, C. Tiozzo, S. Zacchini and P. Zanello, Inorg. Chem., 2007, 46, 7971–7981 CrossRef CAS PubMed.
-
J. L. Atwood and J. W. Steed, in Supramolecular Chemistry, 2nd Edition, 2009, p. 402 Search PubMed.
- S. Bhattacharya and B. K. Saha, CrystEngComm, 2011, 13, 6941–6944 RSC.
- A. J. Close, P. Kemmitt, M. K. Emmerson and J. Spencer, Tetrahedron, 2014, 70, 9125–9131 CrossRef CAS.
- K. Branko and Z. Marija, Acta Chim. Slov., 2012, 59, 670–679 Search PubMed.
- G. T. Tigineh, Y.-S. Wen and L.-K. Liu, Tetrahedron, 2015, 71, 170–175 CrossRef CAS.
- J. Spencer, N. Anjum, H. Patel, R. Rathnam and J. Verma, Synlett, 2007, 2557–2558 CrossRef CAS.
- J. Spencer, C. B. Baltus, H. Patel, N. J. Press, S. K. Callear, L. Male and S. J. Coles, ACS Comb. Sci., 2011, 13, 24–31 CrossRef CAS PubMed.
- C. C. Stoumpos, T. C. Stamatatos, H. Sartzi, O. Roubeau, A. J. Tasiopoulos, V. Nastopoulos, S. J. Teat, G. Christou and S. P. Perlepes, Dalton Trans., 2009, 1004–1015 RSC.
- G. S. Papaefstathiou, C. P. Raptopoulou, A. Tsohos, A. Terzis, E. G. Bakalbassis and S. P. Perlepes, Inorg. Chem., 2000, 39, 4658–4662 CrossRef CAS.
- T. C. Stamatatos, K. A. Abboud, S. P. Perlepes and G. Christou, Dalton Trans., 2007, 3861–3863 RSC.
- G. C. Vlahopoulou, T. C. Stamatatos, V. Psycharis, S. P. Perlepes and G. Christou, Dalton Trans., 2009, 3646–3649 RSC.
- W.-H. Zhu, Y. Zhang, Z. Guo, S. Wang, J. Wang, Y.-L. Huang, L. Liu, Y.-Q. Fan, F. Cao and S.-W. Xiang, RSC Adv., 2014, 4, 49934–49941 RSC.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1066 CrossRef CAS.
- P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS.
- K. Raghavachari and G. W. Trucks, J. Chem. Phys., 1989, 91, 1062–1065 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef.
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS.
-
U. Wedig, M. Dolg, H. Stoll and H. Preuss, Quantum Chemistry: The Challenges of Transition Metals and Coordination Chemistry, Dordrecht, 1986, pp. 79–89 Search PubMed.
- M. F. Summers, Coord. Chem. Rev., 1988, 86, 43–134 CrossRef CAS.
-
M. Frisch, G. Trucks and H. Schlegel, Gaussian Inc., Wallingford, 2010.
-
http://jmol.sourceforge.net/
.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- O. V. Dolomanov, A. J. Blake, N. R. Champness and M. Schröder, J. Appl. Crystallogr., 2003, 36, 1283–1284 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic table of all compounds; ESI-MS of compounds 7, 10 and 16; protocols for thermal and microwave-mediated synthesis of ligands; 1H and 13C NMR of ligands; FT-IR of ligands H2L1–H2L20. CCDC 1406218–1406237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce02109j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*