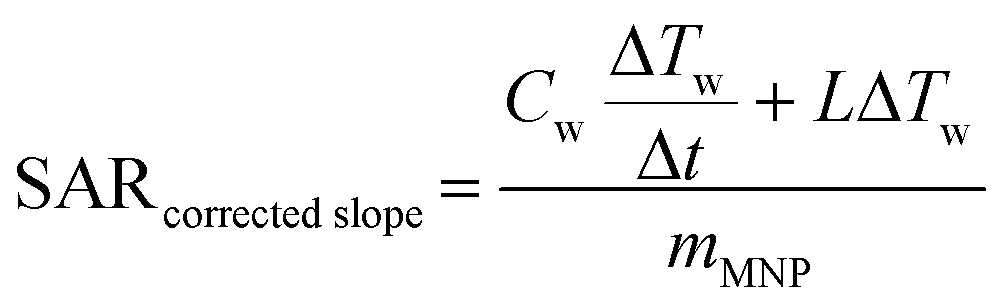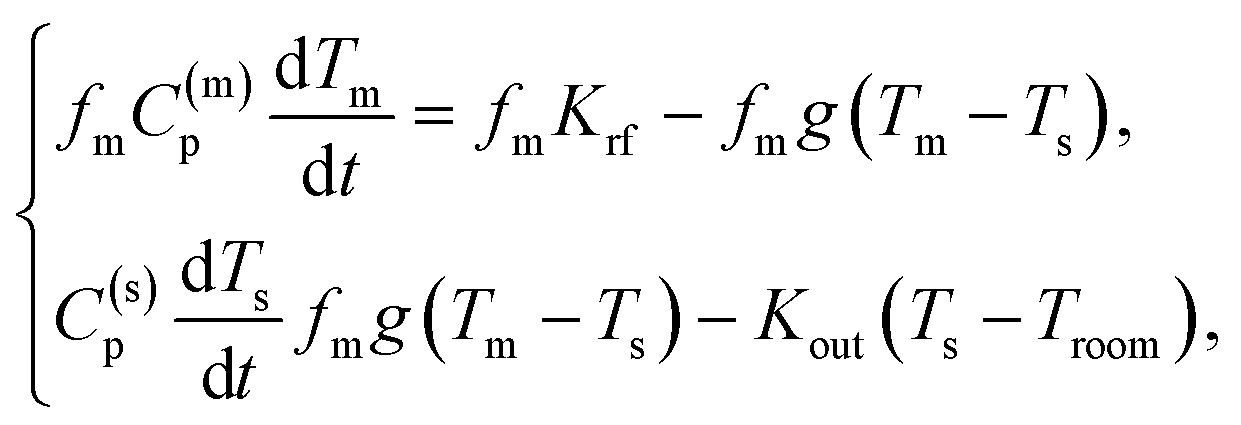DOI:
10.1039/C5CE01890K
(Paper)
CrystEngComm, 2016,
18, 407-416
Evaluation of La1−xSrxMnO3 (0 ≤ x < 0.4) synthesised via a modified sol–gel method as mediators for magnetic fluid hyperthermia†
Received
24th September 2015
, Accepted 27th November 2015
First published on 7th December 2015
Abstract
A range of lanthanum strontium manganates (La1−xSrxMnO3–LSMO) where 0 ≤ x < 0.4 were prepared using a modified peroxide sol–gel synthesis method. The magnetic nanoparticle (MNP) clusters obtained for each of the materials were characterised using scanning electron microscopy (SEM), X-ray powder diffraction (XRD) and infra-red (IR) spectroscopy in order to confirm the crystalline phases, crystallite size and cluster morphology. The magnetic properties of the materials were assessed using the Superconducting quantum interference device (SQUID) to evaluate the magnetic susceptibility, Curie temperature (Tc) and static hysteretic losses. Induction heating experiments also provided an insight into the magnetocaloric effect for each material. The specific absorption rate (SAR) of the materials was evaluated experimentally and via numerical simulations. The magnetic properties and heating data were linked with the crystalline structure to make predictions with respect to the best LSMO composition for mild hyperthermia (41 °C ≤ T ≤ 46 °C). La0.65Sr0.35MnO3, with crystallite diameter of 82.4 nm, (agglomerate size of ∼10 μm), Tc of 89 °C and SAR of 56 W gMn−1 at a concentration 10 mg mL−1 gave the optimal induction heating results (Tmax of 46.7 °C) and was therefore deemed as most suitable for the purposes of mild hyperthermia, vide infra.
Introduction
Magnetic nanoparticles (MNPs) have emerged as promising mediators for localised magnetic hyperthermia treatment and drug delivery.1 The magnetocaloric effect caused by inducing a radio-frequency alternating magnetic field close to an area containing the MNPs leads to a local increase in temperature. When this local increase in temperature occurs within the range of 41–46 °C it is known as mild hyperthermia.2 Mild hyperthermia is a promising therapy at the cellular3 and systemic levels, allowing the selective targeting of cancer cells through the exploitation of the characteristic traits of cancer cells whilst minimising the damage to healthy tissue.4–6 The therapeutic use of mild hyperthermia is viewed as an adjunct to conventional chemo- or radio- therapies, allowing them to be more effective and require lower dosages as a result. For MNPs to be considered for mild hyperthermia they should be biocompatible; have a low Curie temperature (Tc), ideally within or close to the therapeutic range of mild hyperthermia (41–46 °C); and have a high Specific Absorption Rate (SAR).7 SAR is a measure of the ability of a material to absorb energy when exposed to electromagnetic radiation.
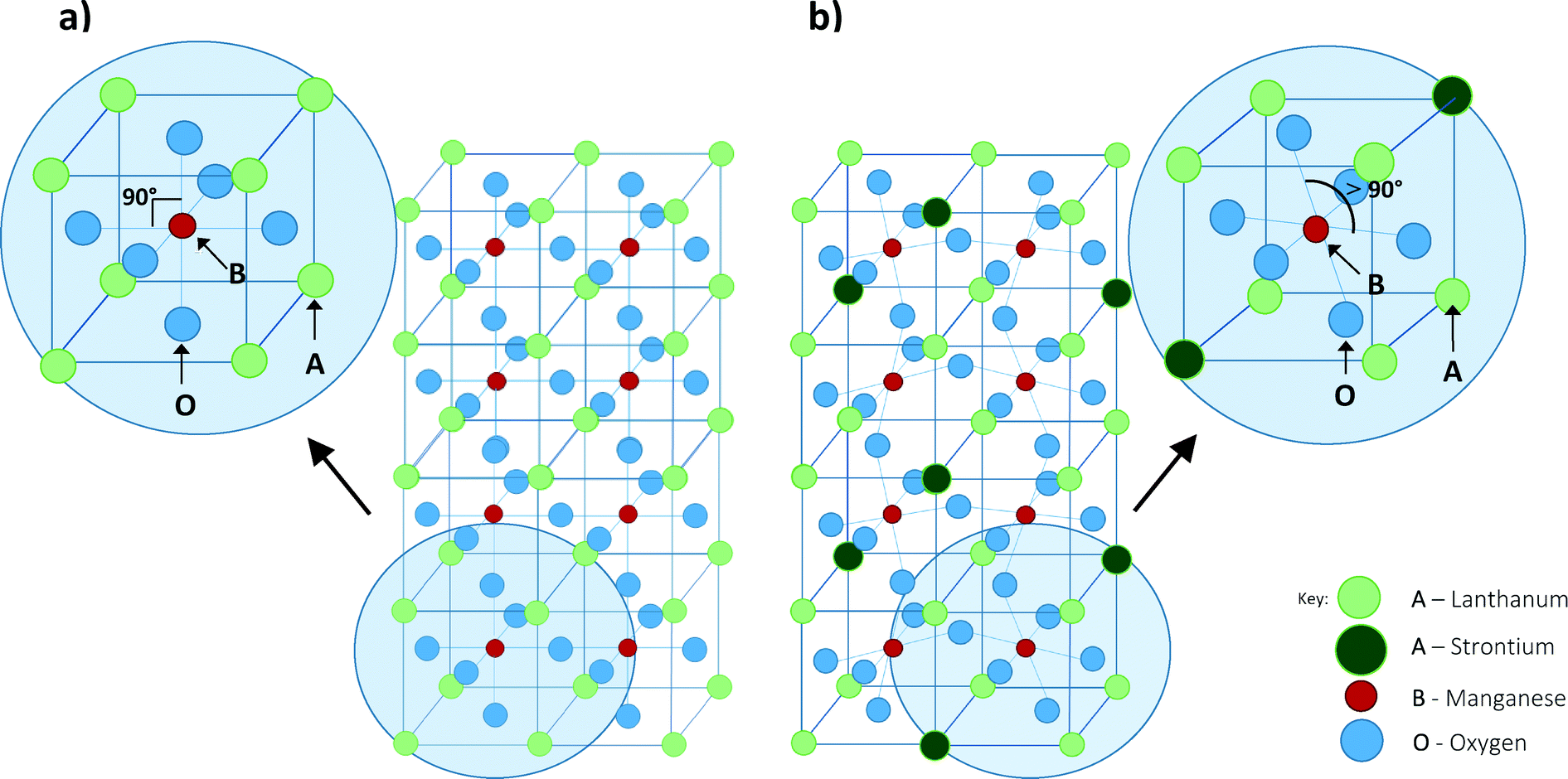
The vast majority of magnetic nanoparticles investigated for hyperthermia treatment until recently were iron-based8–15 which are biocompatible and have high SARs16 (of the order of 100 W g−1). However, iron-based materials have high Tcs (ranging from 280 °C (ref. 17) to 956 °C (ref. 18)) meaning that application of the magnetic field may lead to overheating of local tissue.19 This issue has led to a variety of doped composite materials being developed such as ferrites of Pd–Ni,7 Ni–Cu,20 Co–Au (ref. 21) and Co–Zn.22,23 The aim of these studies was to reduce the Tc to within the therapeutic range so as to reduce the likelihood of overheating, whilst optimising the SAR. Prior research into perovskite-based manganate MNPs17,24–30 also shows them to be promising materials. The potential of these materials as mediators for hyperthermia relies on their perovskite structure. Their composition and as such, magnetic properties, can be tailored without significantly changing the perovskite phase. This can occur by simply distorting the crystal, without compromising the stability. Regardless of the crystal structure the resulting material remains single phase as opposed to composite core–shell materials whose stability may be less predictable. Fig. 1 illustrates the perovskite structure ABO3 present in the manganates. Perovskites have a face-centred cubic lattice with A-sites found on the corners, and the B-sites in the centre of the unit cells. The magnetic properties of a perovskite, including the transition temperature, can be tailored by doping the A and/or B site ions.31,32 Altering the magnetic properties of the material may also be achieved by means of altering the method of synthesis. For example, increasing the sintering temperature for La0.7Sr0.3MnO3 from 600–1000 °C leads to an increase in the grain size from 37 to 163 nm and an increase in Tc from 38–57 °C.33
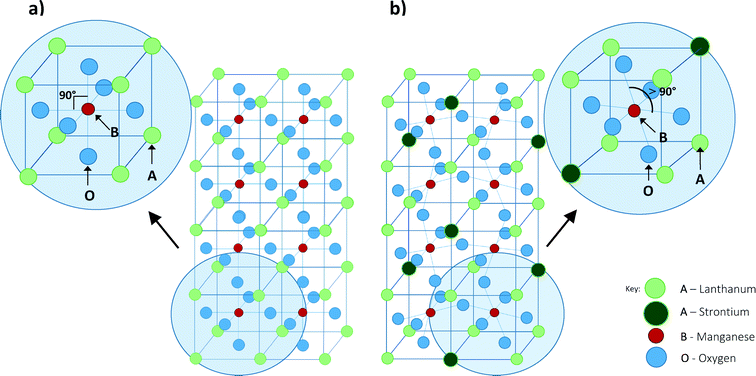 |
| | Fig. 1 Structure of perovskites of the type ABO3. Fig. 1a shows the undoped parent compound LaMnO3 and Fig. 1b shows the distortion caused to the lattice with dopant in La1−xSrxMnO3 compounds. | |
In this work we have investigated the effect of dopant concentration on the magnetic properties (magnetic susceptibility, SAR and Tc) of lanthanum strontium manganates (La1−xSrxMnO3) prepared using a modified sol–gel method. This perovskite family has been suggested as a candidate material for MNP mediated hyperthermia treatment.6 Previous studies have investigated the effect of the particle size33 and the effect of altering the composition32,34,35 on the magnetic properties of the La1−xSrxMnO3 (LSMO) MNPs. As composition is varied with dopant, a change in magnetic properties is observed. This paper presents a systematic study of the changes in structure, magnetic susceptibility, hysteresis as well as magnetocaloric effect as a result of the changes in composition. The simultaneous investigation of structural changes and changes to physical properties is not usually seen in the literature.
The La1−xSrxMnO3 family is a group of mixed metal oxides of the perovskite structure where La3+ or Sr2+ ions are the A-site cations with either Mn3+ or Mn4+ as the B-site cation. The substitution of La3+ ions for larger Sr2+ ions causes distortion of the local crystal lattice away from the ideal cubic geometry of the parent compound through orthorhombic and rhombohedral structures. This alters the chemical pressure exerted within the lattice, and so alters the bond length of Mn–O and the Mn3+–O2−–Mn4+ bond angle. Where this bond angle nears 180° and with decreasing bond length, the extent of eg orbital overlap increases leading to enhancement of the double-exchange mechanism.36 The double-exchange mechanism is a type of magnetic exchange whereby an electron is simultaneously transferred from Mn3+ to O2− while an electron is transferred from O2− to Mn4+. Enhancement of the double-exchange mechanism leads to enhancement of magnetic properties such as magnetoresistance and the magnetocaloric effect.36
The dopant ion may also be used to affect the valency of the B-site cation. For example in La1−xSrxMnO3 with small levels of Sr2+ dopant, the ratio of La3+ to Sr2+ decreases and so the ratio of Mn4+ to Mn3+ increases. Increasing the proportion of Mn4+ towards a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio favours the double exchange mechanism.37 Mn4+ is known to be a non Jahn–Teller active ion because its three valence electrons occupy degenerate t2g orbitals, whereas Mn3+ is Jahn–Teller active due to its four valence electrons in a high spin state occupying non-degenerate orbitals.38 This degeneracy causes distortion of the bond lengths in order to minimise the energy of conformation. Therefore, Jahn–Teller distortion will be less effective at increasing the Mn3+–O2−–Mn4+ bond angle to 180° with higher levels of Sr2+/Mn4+. This would lead to the restriction of the double exchange mechanism, and hence the magnetocaloric effect would be limited. This correlates to the fact that LSMO in the entire range of 0.1 < x < 0.5 is in the ferromagnetic phase.37
:1 ratio favours the double exchange mechanism.37 Mn4+ is known to be a non Jahn–Teller active ion because its three valence electrons occupy degenerate t2g orbitals, whereas Mn3+ is Jahn–Teller active due to its four valence electrons in a high spin state occupying non-degenerate orbitals.38 This degeneracy causes distortion of the bond lengths in order to minimise the energy of conformation. Therefore, Jahn–Teller distortion will be less effective at increasing the Mn3+–O2−–Mn4+ bond angle to 180° with higher levels of Sr2+/Mn4+. This would lead to the restriction of the double exchange mechanism, and hence the magnetocaloric effect would be limited. This correlates to the fact that LSMO in the entire range of 0.1 < x < 0.5 is in the ferromagnetic phase.37
LSMO is being considered here as a mediator for hyperthermia treatment as this family of materials shows the most promise to tune the Tc within the desired range by varying the level of strontium dopant.39 In preliminary testing, the maximum Tc recorded for La0.75Sr0.25MnO3 was 98 °C (60 nm)40 compared with Fe3O4 for which 585 °C (100 nm) was observed.26 Hence there is more potential to reduce the Tc to within therapeutically acceptable levels using manganates by means of altering the preparation method or composition of the material through doping.31,32 The method of preparation adopted in this work is a modified peroxide sol–gel synthesis41 which has not been used to synthesise these materials before for the application as mediators for hyperthermia treatment. This facile method of synthesis uses metal oxide and metal carbonate precursors in water, along with hydrogen peroxide and a small amount of ammonium hydroxide. The Pechini42 method has been widely used in this area instead as it uses metal nitrate precursors which are extremely soluble and so form a very stable solution. Upon gelation and aging of this solution it is possible to synthesise highly monodisperse and uniform nanoparticles. The modified peroxide sol–gel method was used here as it avoids the use of EDTA and citric acid as the chelating agents, using hydrogen peroxide instead which leads to water as the only by-product of the reaction. It is deemed to be a safer method of preparation in the lab scale as it does not require the use of highly oxidising nitrate precursors. Further investigation in order to achieve better control of the particle size and particle size distribution is currently underway.
Experimental
Material synthesis
As mentioned previously an aqueous modified sol–gel method was used in this work based on a method developed by Liu et al.43 and Chen et al.44 The powder precursors (obtained from Sigma Aldrich); lanthanum(III) oxide, ≥99.9% (La2O3); manganese(II) carbonate, ≥99.9 % trace metals basis; and strontium carbonate, ≥99.9 % trace metals basis; were added to 150 ml deionised H2O in appropriate proportions in order to obtain the desired stoichiometric ratio for the different Sr-doping levels. No thermal pre-treatments were carried out on the powders. While stirring, 100 mL of hydrogen peroxide (30 vol% – Fisher Scientific) was added slowly, followed by addition of 3 ml of ammonium hydroxide NH4OH (25 vol%, Sigma Aldrich). Hydrogen peroxide worked as a complexation agent in addition to participating in the oxidation of manganese, while the role of ammonia was to peptize the solution into a stable solution. Chen et al. found that the addition of hydrogen peroxide was necessary in order to obtain a stable solution and xerogel which is why a larger excess of peroxide is used than is needed for simply the oxidation of Mn2+ to Mn3+ and Mn4+.44
After stirring at 70 °C for 2 hours, the resulting solution was dried at 90 °C until dried gel formation. Finally calcination at 1100 °C for 16 hours was used to remove the residual carbon and obtain a crystalline product. After calcination, the xerogel was converted into a powder which contained the three components in the desired stoichiometric level in the perovskite phase. Table 1 shows all the produced samples and sample names to be used for the remainder of the report. The samples were manually ground before characterisation and testing.
Table 1 Lattice parameters and atomic coordinates for (a) orthorhombic (Pnma) and (b) rhombohedral (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) which were used to as input parameters for Rietveld analysis
c) which were used to as input parameters for Rietveld analysis
| (a) Orthorhombic (Pnma) lattice parameters and atomic coordinates a = 5.5743, b = 7.695 and c = 5.537 |
| Atom |
Wycoff position |
x
|
y
|
z
|
| La |
4c |
0.55 |
0.3 |
0 |
| Mn |
4a |
0 |
0 |
0 |
| O(1) |
4c |
−0.011 |
0.3 |
−0.1 |
| O(2) |
8d |
0.309 |
0 |
0.2 |
| (b) Rhombohedral (Rc) lattice parameters and atomic coordinates a = b = 5.5212 and c = 13.37908 |
| Atom |
Wycoff position |
x
|
y
|
z
|
| La |
6a |
0 |
0 |
0.3 |
| Mn |
6b |
0 |
0 |
0 |
| O |
18e |
0.55 |
0 |
0.3 |
Characterisation
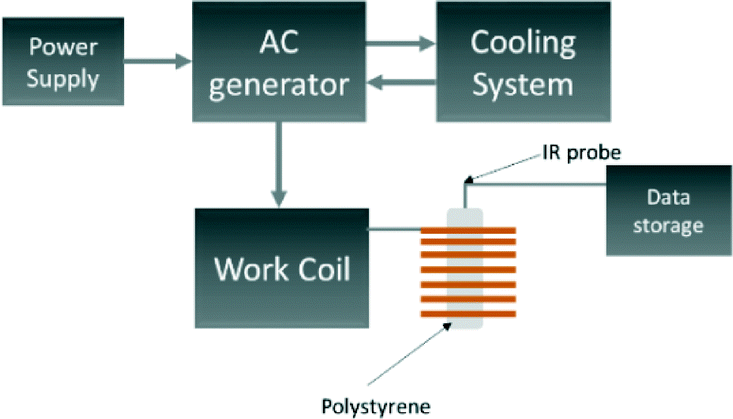
The produced samples underwent a series of analysis in order to obtain the necessary information with respect to the crystalline structure, sample morphology and magnetic properties. Structural determination was carried out using XRD (Panalytical X'Pert) and Fourier Transform Infrared Spectroscopy (FTIR Perkin Elmer Spectrum 2). The Scherrer equation using full-width at half maximum was used to determine the size of the crystallites using the most intense reflection in all cases, which occurred at approximately 32 degrees 2θ. As the Bragg angle is obtained to 4 decimal places, the error propagation of the trigonometric Scherrer function comes to a value of ±0.1 nm in keeping with the values quoted. X'Pert High Score Plus was used to carry out a Rietveld analysis on the diffraction patterns of the crystalline phases present in the powder samples. The input lattice parameters and atomic coordinates which best matched the experimental data are shown in Table 1a and b. The background was not subtracted and refinement of the atomic coordinates of both phases was undertaken before refining the instrumental parameters. The morphology of the resulting samples was studied using SEM (eSEM – FEI Quanta FEG – Environmental SEM Oxford Ex-ACT). The concentration of dopants in the synthesised materials was then confirmed using eSEM-EDX with the Aztec based XACT system. Atomic percentage was determined using the relative ratio of La to Sr elements in each sample. A cobalt standard was used with the uncertainty for the data values at the ±1% level. The magnetic susceptibility and magnetisation hysteresis were measured using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS XL). Field dependent magnetisation measurements were conducted in the range −10000 ≤ H ≤ 10000 Oe, at 100 K and 200 K. Susceptibility measurements were conducted in field cooled (FC) and zero field cooled (ZFC) conditions between 100 and 370 K, at 500 Oe. Non-adiabatic heating experiments were conducted using an RF magnetic field generator (Easyheat 0112) coupled with a water-cooled induction coil (6-turn 12 cm length coil).
A schematic of the induction rig used for the magnetic heating measurements is shown in Fig. 2. The generated magnetic field characteristics were frequency: 175 kHz and amplitude: 10.95 kAm−1. Aqueous suspensions of the MNPs of concentrations 5, 10 and 15 mg mL−1 were prepared. The temperature of the aqueous suspension during the induction heating experiments was measured with an IR temperature probe (Optocon AG) FOTEMP 1 coupled to a (Optocon AG) TS2/2 sensor. The heating curves from the induction heating experiments are presented in terms of temperature difference rather than measured temperature to reduce systematic errors. The results obtained from the induction heating measurements were used to calculate the SAR. The corrected slope method was used here to ensure reliable results which can be compared between different experimental set-ups. The SAR was estimated using the following equation:45
| | 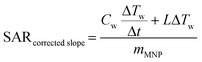 | (1) |
where
Cw is the specific heat capacity (J g
−1 K
−1) of water,
mMNP is the weight fraction of the magnetically active element, Δ
Tw/Δ
t is the change in temperature against time, and
L is the thermal losses of the system (taken from the literature
45).
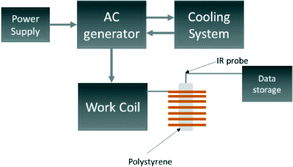 |
| | Fig. 2 Schematic of equipment used for the induction heating experiments. | |
It has been recently reported that a temperature increase (3 °C in a 10 minute period) may be observed without a magnetic sample present inside the induction coil. This is a result of large currents flowing through the induction coil causing additional convectional heating.45 As a result, the temperature of 1 mL of water was also measured under the same conditions to act as a blank experiment in order to correctly attribute heating effects as being due to the magnetocaloric effect of the material. We found that for our experimental settings, the temperature of the blank reproducibly increased by 3 °C in a 10 minute period when the initial temperature was 25 °C, in agreement with the literature. The magnetic heating curves here are not scaled to remove the blank heating profile as they would then not be comparable to other published results. However, the blank is taken into consideration when calculating the linear loss parameter for the SAR using Wildeboer et al.'s method.45 We use the SAR of the blank in Fig. 8 and 9 as a guide to compare SARs for the range of materials.
The experimental heating curves were modelled using a simple two-temperature model.46 This model is based on the balance between: (i) the energy absorbed by the MNP; (ii) the energy released by the MNP to the solvent; (iii) the heat loss to the environment. This energy balance can be written down as a system of two ordinary differential equations:
where
Tm is the temperature of the MNP,
Ts is the temperature of the solvent,
Troom is the temperature of the environment,
C(m)p is the specific heat capacity at constant pressure of the MNP,
C(s)p is the specific heat capacity at constant pressure of the solvent,
fm is the sample concentration of MNPs in water,
g is specific heat transfer coefficient,
Krf is the specific absorption rate of the MNPs (and not per g
Mn as is the SAR), and
Kout is the specific loss rate of the solvent to the environment. To simplify the calculations, we assume that all the parameters in the equations are temperature independent. In particular, we set
C(m)p = 1.44706 × 10
3 J kg
−1 K
−1 and
C(s)p = 42 × 10
3 J kg
−1 K
−1 (for water). The sample concentration has been set to
f = 10 mg g
−1 = 0.01.
Results and discussion
Crystalline structure and configuration analysis
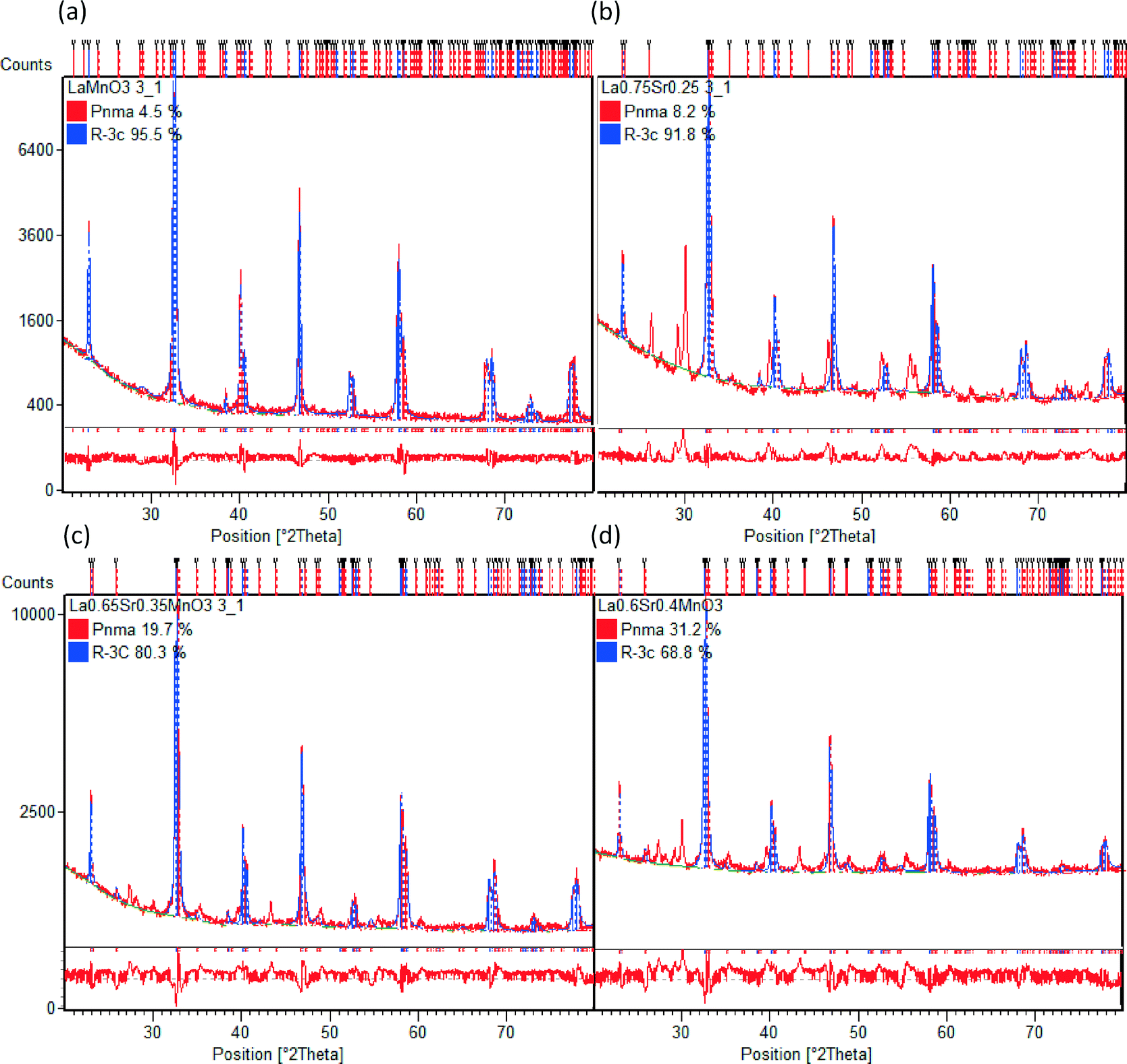
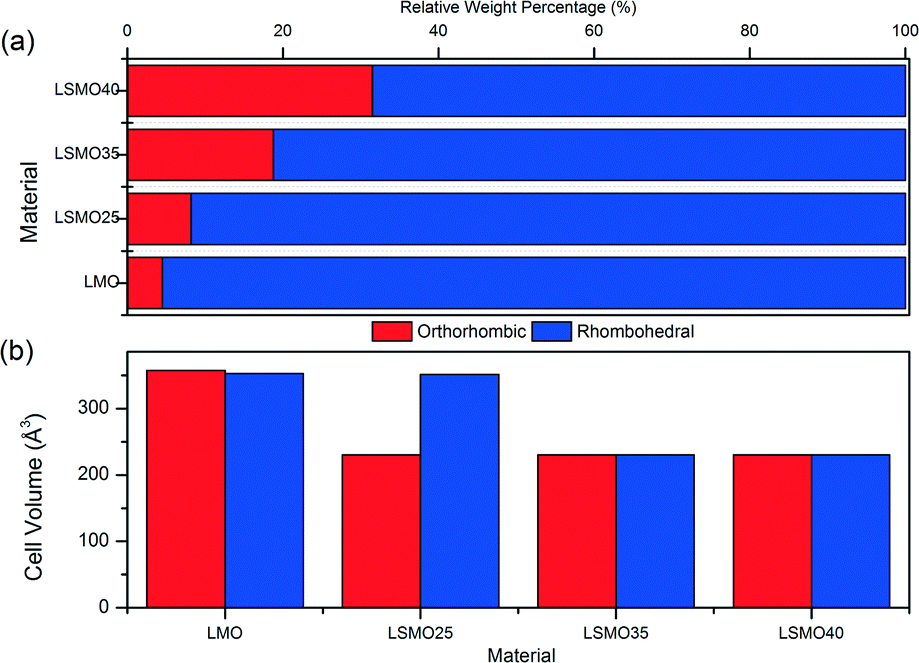
Table 2 shows the stoichiometry of the synthesised samples as verified by EDX to be in keeping with the desired ratios (the EDX maps of all samples are provided in the ESI†). In Fig. 3 the XRD patterns for the LSMO samples can be seen. An increasing proportion of the secondary phase of SrMnO3 is visible with increasing strontium dopant. Also evident is the increasing intensity of the reflections and (110) in particular, with increasing strontium dopant. Peaks at 28–30° 2θ are likely to be due to contamination with agate during manual grinding. Other potential secondary phases which may be present in even smaller amounts include La2O3, MnCO3 and SrMnO3.47 Previous observations by Zhang et al. have shown that for x < 0.2 the orthorhombic geometry is typically dominant, followed by a transition to rhombohedral geometry for 0.25 < x < 0.5.48 On initial assessment of the diffraction patterns our data seemed to support this but the presence of more than one phase is evident in the shallow transition in magnetic susceptibility vide infra. On completion of Rietveld analysis of the samples we found that all samples produced via this modified peroxide sol–gel method contained mixed crystalline phases. The calculated crystalline phases which best fitted the experimental data were found to be orthorhombic (Pnma)49 and rhombohedral (Rc)50 crystal structures. The rhombohedral (Rc) geometry appears to be the dominant crystalline phase for all the samples presented here (LMO, LSMO25, LSMO35 and LSMO40) in keeping with literature.51–53Fig. 4 shows that with increasing strontium dopant, the relative weight percentage of the orthorhombic (Pnma) crystalline phase increases. This is in keeping with Dokiya et al. who documented the structural transition of LSMO from rhombohedral (Rc) to higher orthorhombic and rhombohedral symmetries with increasing strontium dopant or decreasing oxygen content.49
Table 2 Key characterisations on samples presented in this work including the stoichiometry of each sample as determined by EDX analysis, as well as the average crystallite diameter calculating using the Scherrer equation using the most intense peak
| Sample |
Sample name |
EDX |
XRD |
IR |
Induction heating |
SEM |
M vs. T (SQUID) |
M V H (SQUID) |
EDX stoichiometry |
Crystallite diameter (nm) |
| LaMnO3 |
LMO |
|
✓ |
✓ |
✓ |
|
|
|
— |
164.9 |
| La0.8Sr0.2MnO3 |
LSMO20 |
✓ |
✓ |
✓ |
✓ |
|
|
|
La0.82Sr0.18MnO3 |
82.4 |
| La0.75Sr0.25MnO3 |
LSMO25 |
✓ |
✓ |
✓ |
✓ |
|
|
|
La0.76Sr0.24MnO3 |
82.4 |
| La0.7Sr0.3MnO3 |
LSMO30 |
✓ |
✓ |
✓ |
✓ |
|
✓ |
✓ |
La0.72Sr0.28MnO3 |
82.4 |
| La0.65Sr0.35MnO3 |
LSMO35 |
✓ |
✓ |
✓ |
✓ |
|
✓ |
✓ |
La0.66Sr0.34MnO3 |
82.4 |
| La0.6Sr0.4MnO3 |
LSMO40 |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
La0.62Sr0.38MnO3 |
61.9 |
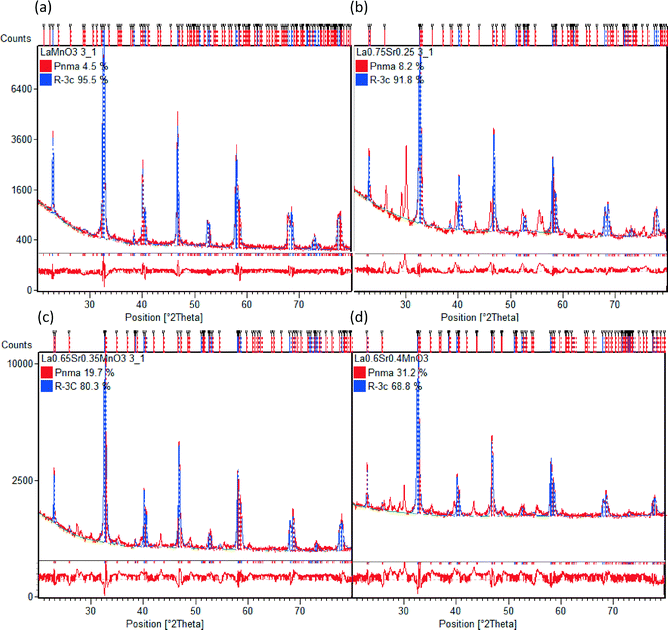 |
| | Fig. 3 Rietveld analyses of X-ray diffraction patterns of a selection of LSMO materials (a) LMO (b) LSMO25 (c) LSMO35 and (d) LSMO40 to quantify the relative weight percentages of orthorhombic (Pnma) and rhombohedral (Rc) crystalline phases. The red line below each diffraction pattern is the difference plot which gives an indication of the variation between the calculated and experimental data. | |
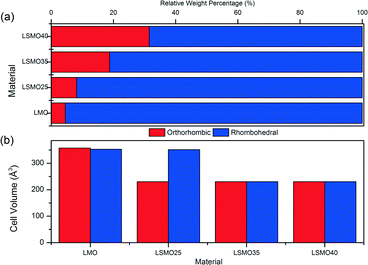 |
| | Fig. 4 Crystal structure comparison of orthorhombic and rhombohedral phases for (a) relative weight percentages and (b) unit cell volumes for LMO, LSMO25, LSMO35 and LSMO40. | |
Table 3 shows the calculated structural information for the LSMO samples. We can see that the A-site substitution of lanthanum with cationic radius of 1.36 Å for strontium with a larger cationic radius of 1.44 Å does not account for the decrease in cell volume with increasing dopant concentration for both phases. The decrease in cell volume is therefore ascribed to the decreasing ionic radii of the B-site with increasing strontium dopant, which causes Mn3+ with cationic radii of 0.645 Å to be replaced by the smaller Mn4+ with cationic radii of 0.53 Å.54 There appears to be little difference in cell volumes for orthorhombic and rhombohedral crystalline phases in each of the LMO, LSMO35 and LSMO40 samples. For LSMO25 however, the cell volume of the rhombohedral structure is 52% greater than for the orthorhombic structure. It is at this point that the relative weight percentage of the rhombohedral phase begins to more rapidly decline, with the orthorhombic phase more rapidly increasing. For both crystal phases we can see a reduction in cell volume of approximately 35% between x = 0 and x = 0.35. It is in this range that we could expect the material to have enhanced magnetic properties as the decrease in cell volume would lead to shorter bond lengths with greater bonding orbital overlap meaning the double exchange mechanism will be more effective. The change in crystal structure from rhombohedral to orthorhombic geometry also means that γ has decreased from 120–90°. In doing so this should mean that a greater proportion of Mn3+–O–Mn4+ bonds are close to 180° for optimal double exchange.36 By this rationale LSMO35 and LSMO40 should be able to participate more effectively in the double exchange mechanism and therefore exhibit better magnetocaloric effect. The SEM analysis shown in Fig. 5 (LSMO40) indicates that the samples consist of agglomerates of nanoparticles of the order of 10 μm.
Table 3 Refined lattice parameters resulting from Rietveld analysis for LMO, LSMO25, LSMO35 and LSMO40 (Gof – goodness of fit)
| Sample |
Crystal system |
a/Å |
b/Å |
c/Å |
α/° |
β/° |
γ/° |
V/Å |
Space group |
R
w
|
R
exp
|
R
bragg
|
Gof |
Wt percent./% |
| LMO |
Orthorhombic |
7.43101 |
8.4159 |
5.7144 |
90 |
90 |
90 |
357.4 |
Pnma
|
9.4 |
4.1 |
40.3 |
5.1 |
4.5 |
| Rhombohedral |
5.52248 |
5.5225 |
13.3686 |
90 |
90 |
120 |
353.1 |
Rc |
6.2 |
95.5 |
| LSMO25 |
Orthorhombic |
5.43725 |
7.7984 |
5.43306 |
90 |
90 |
90 |
230.4 |
Pnma
|
10.7 |
3.5 |
4.22 |
9.3 |
8.2 |
| Rhombohedral |
5.51461 |
5.5146 |
13.3552 |
90 |
90 |
120 |
351.7 |
Rc |
2.64 |
91.8 |
| LSMO35 |
Orthorhombic |
5.44815 |
7.7433 |
5.46776 |
90 |
90 |
90 |
230.4 |
Pnma
|
8.2 |
3.8 |
18.8 |
4.6 |
19.7 |
| Rhombohedral |
5.51197 |
5.5119 |
13.3546 |
90 |
90 |
120 |
351.4 |
Rc |
28.8 |
80.3 |
| LSMO40 |
Orthorhombic |
5.44819 |
7.7501 |
5.45624 |
90 |
90 |
90 |
230.4 |
Pnma
|
6.1 |
2.7 |
29.45 |
5.03 |
31.2 |
| Rhombohedral |
5.51086 |
5.5109 |
13.3543 |
90 |
90 |
120 |
351.2 |
Rc |
9.57 |
68.8 |
 |
| | Fig. 5 Scanning electron micrograph of LSMO40. | |
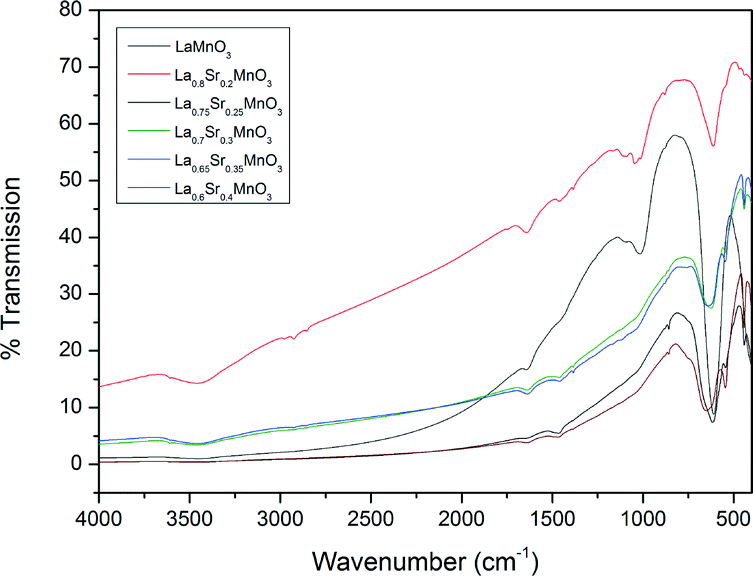
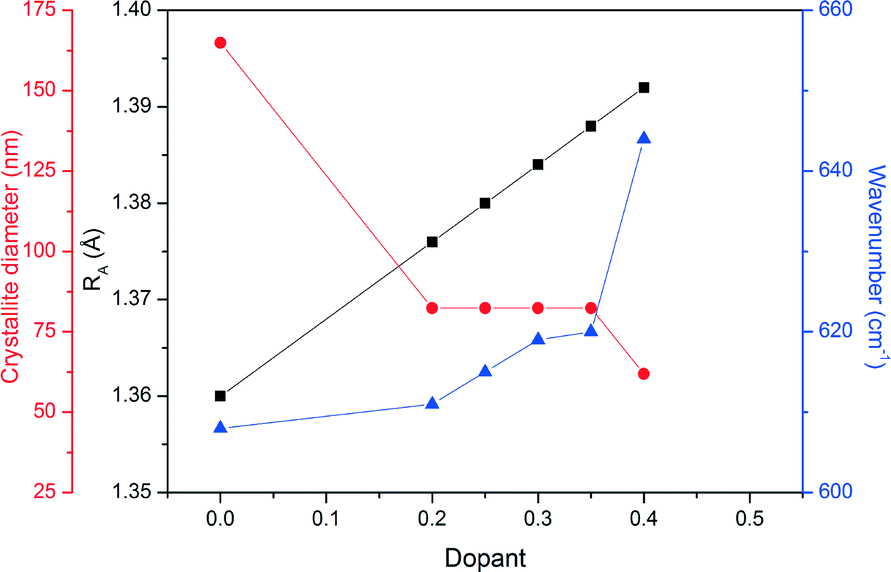
Fig. 6 shows the FTIR analysis results for the prepared LSMO samples. The presence of octahedral MnO6 due to the absorption at approximately 600 cm−1 by the Mn–O bond can be seen in all cases. However, there is a positive shift in the wavenumber of this absorption with increasing dopant concentration. Fig. 7 depicts the calculated parameters from the XRD and FTIR analysis (crystallite size, average A-site cation radius and Mn–O bond absorption wavenumber) as a function of the dopant concentration. We can see that increasing the dopant concentration (and as a result the average radius of the A-site cation) decreases the crystallite diameter for LSMO and in turn shortens the bond angle β (shown in Table 3) until a geometrical transition (from rhombohedral to orthorhombic) is observed while increasing the Mn–O wavenumber. This increase in wavenumber is evidence of the increasing energy required to manipulate the Mn–O bond in the more distorted octahedron where the geometric transition has occurred to reduce strain. For LSMO samples where 0.2 < x < 0.35 the crystallite diameter remains constant, yet increasing strain caused by increasing dopant concentration creates greater internal pressure in the perovskite lattice. This internal pressure, based on this rationale is at its maximum for LSMO35, where we begin to see a significant change in the relative weight percentages of the orthorhombic and rhombohedral geometries. As it is in a region of magnetic phase transition in which the optimal magnetocaloric effect may be observed, we may expect the maximum caloric effect for the LSMO35 sample. This hypothesis will be evaluated next when the magnetisation and induction heating experiments are discussed.
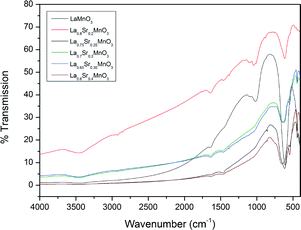 |
| | Fig. 6 IR spectra for a range of the doped LSMO materials where x = 0.2 to 0.4 compared to the parent compound LaMnO3. | |
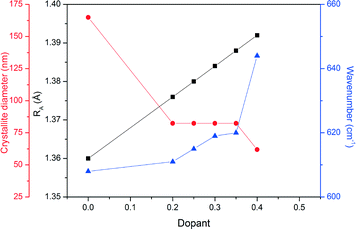 |
| | Fig. 7 The relationship between the average radii of A-site cations (RA), crystallite diameter and wavenumber for the range of doped LSMO materials. Black squares correspond to RA, red circles to the crystallite diameter, and blue triangles to the Mn–O wavenumber. Connecting lines are included as a guide to the eye. | |
Magnetic heating experiments
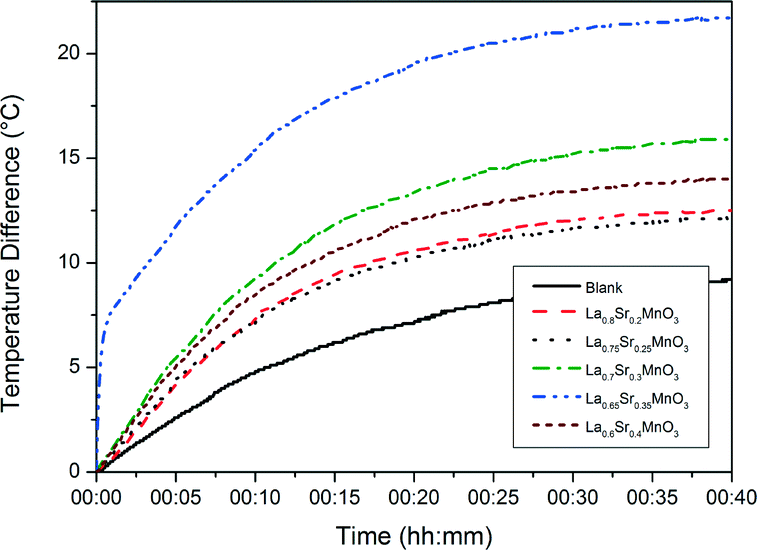
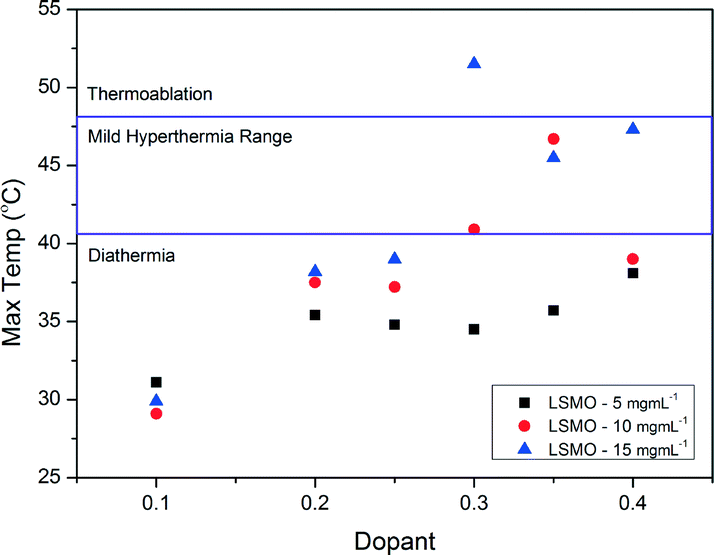
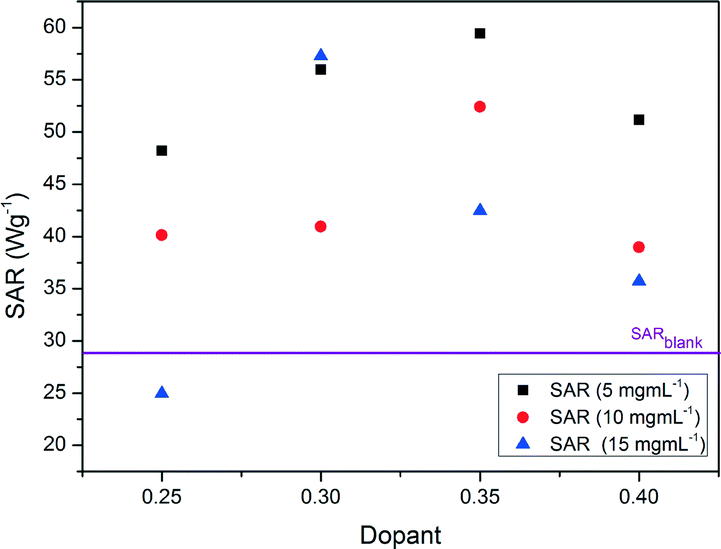
Fig. 8 shows the heating curves for doped LSMO samples where 0.2 < x < 0.35 for sample concentration of 10 mg mL−1; we can see that as dopant increases so does the maximum temperature reached by each heating profile until an optimum is reached, after which this maximum temperature of saturation decreases again. Fig. 9 shows the saturation temperature achieved for all LSMO samples at the three different concentrations (5 mg mL−1, 10 mg mL−1 or 15 mg mL−1); in general increasing the suspension concentration increases the maximum temperature achieved. For example; where x = 0.3, the maximum temperature reached increases from 34.5 °C at 5 mg mL−1, to 40.9 °C at 10 mg mL−1, and to 51.5 °C at 15 mg mL−1. The dopant concentration for obtaining the optimal maximum temperature is also a function of the suspension concentration dropping from x = 0.4 for 5 mg mL−1 (38.1 °C) to x = 0.35 for 10 mg mL−1 (46.7 °C) and to 0.3 for 15 mg mL−1 (51.5 °C). The temperatures of both the blank sample and the parent compound LaMnO3 increase with time in the AC field by similar amounts. In Fig. 10 we can see that the optimal SARs at concentrations of 5, 10 and 15 mg mL−1 are found for samples where 0.3 < x < 0.35. SARs are higher for the range of doped samples when tested at lower concentrations. As SAR is calculated per gram of the magnetic material, the observed decrease in SAR is expected with increasing sample concentration. The greatest optimal SAR of 59.43 W gMn−1 was obtained at a concentration of 5 mg mL−1 where x = 0.35. However the maximum temperature achieved by this sample concentration was 35.7 °C. Eqn (2) and (3) have also been solved numerically and the result for Ts(t) has been fitted to the experimental heating values using a standard least squares regression. The calculated parameters (as discussed previously) as a function of the doping, x, are shown in Table 4. These are in very good agreement with the SARs calculated using Wildeboer et al.'s method45 shown in Fig. 10.
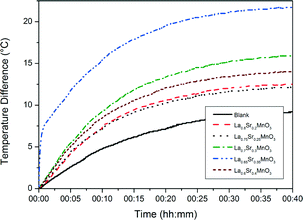 |
| | Fig. 8 Heating curves of the range of doped LSMO materials using aqueous suspension of 10 mg mL−1. | |
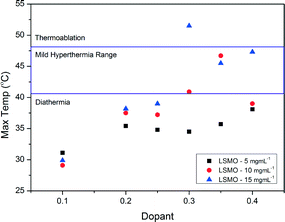 |
| | Fig. 9 Maximum temperatures reached by the range of doped LSMO materials at various suspension concentrations. | |
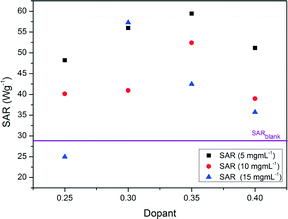 |
| | Fig. 10 Specific absorption rates reached by the range of doped LSMO materials. | |
Table 4 Calculated modelling parameters
|
x
|
K
rf W kg−1 |
g W kg−1 K−1 |
K
out W kg−1 K−1 |
SAR W gMn−1 |
T
max (°C) |
| 0.20 |
12733 |
56.66 |
6.006 |
53.67 |
35.60 |
| 0.25 |
11507 |
50.60 |
5.684 |
47.97 |
35.12 |
| 0.30 |
12888 |
124.01 |
6.706 |
53.12 |
34.61 |
| 0.40 |
13210 |
104.65 |
6.052 |
53.83 |
35.91 |
For the efficacy of the hyperthermia treatment as previously stated, the heat generated should be within the mild hyperthermia range of 41–46 °C with a high SAR. Based on these two considerations, further work was done using LSMO where x = 0.35 and a suspension concentration of 10 mg mL−1 in order to achieve a maximum temperature of 46.7 °C at 10 mg mL−1.
Magnetic measurements
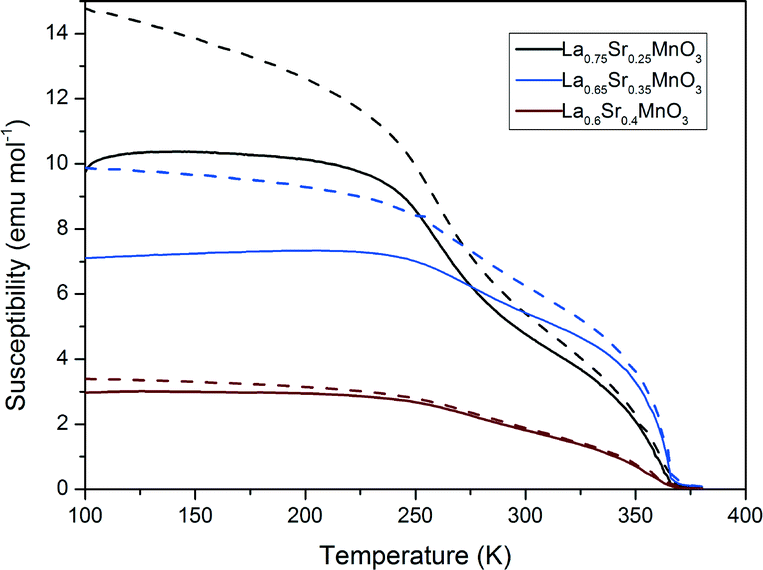
Fig. 11 shows the magnetic susceptibility of the materials with respect to temperature. All samples show a ferromagnetic to paramagnetic transition where the zero-field cooled and field cooled curves overlap. The susceptibilities of the LSMO materials in Fig. 11 decrease with increasing dopant from x = 0.25 to x = 0.4. These LSMO materials have a shallow transition region, exhibiting multiplicity as two slight inflections are observed on each graph. This indicates that the LSMO samples contain more than one magnetic phase present. Comparing the susceptibility measurements with the magnetic heating results we can see that the materials with lower susceptibilities produce a greater magnetocaloric effect and vice versa. We would expect to see that since, as the susceptibility of the material decreases, the A.C. hysteretic loss contributions in particular would increase (vide infra).
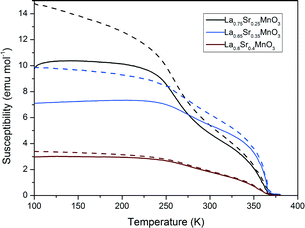 |
| | Fig. 11 Magnetic susceptibly measurements for a selection of LSMO samples (dashed lines represent ZFC and full lines FC measurements). | |
Table 5 shows the corresponding Tcs which were calculated from the susceptibility measurements. The Tc was estimated for each sample using the Arrott plot method. Increasing from x = 0.25 to 0.35 for LSMO, we see that Tc increases and then decreases again where x = 0.40. The sample where x = 0.4 has a higher Tc than the phase diagram would suggest.37 However all calculated Tcs are in keeping with experimental trends reported by Urushibara et al.40
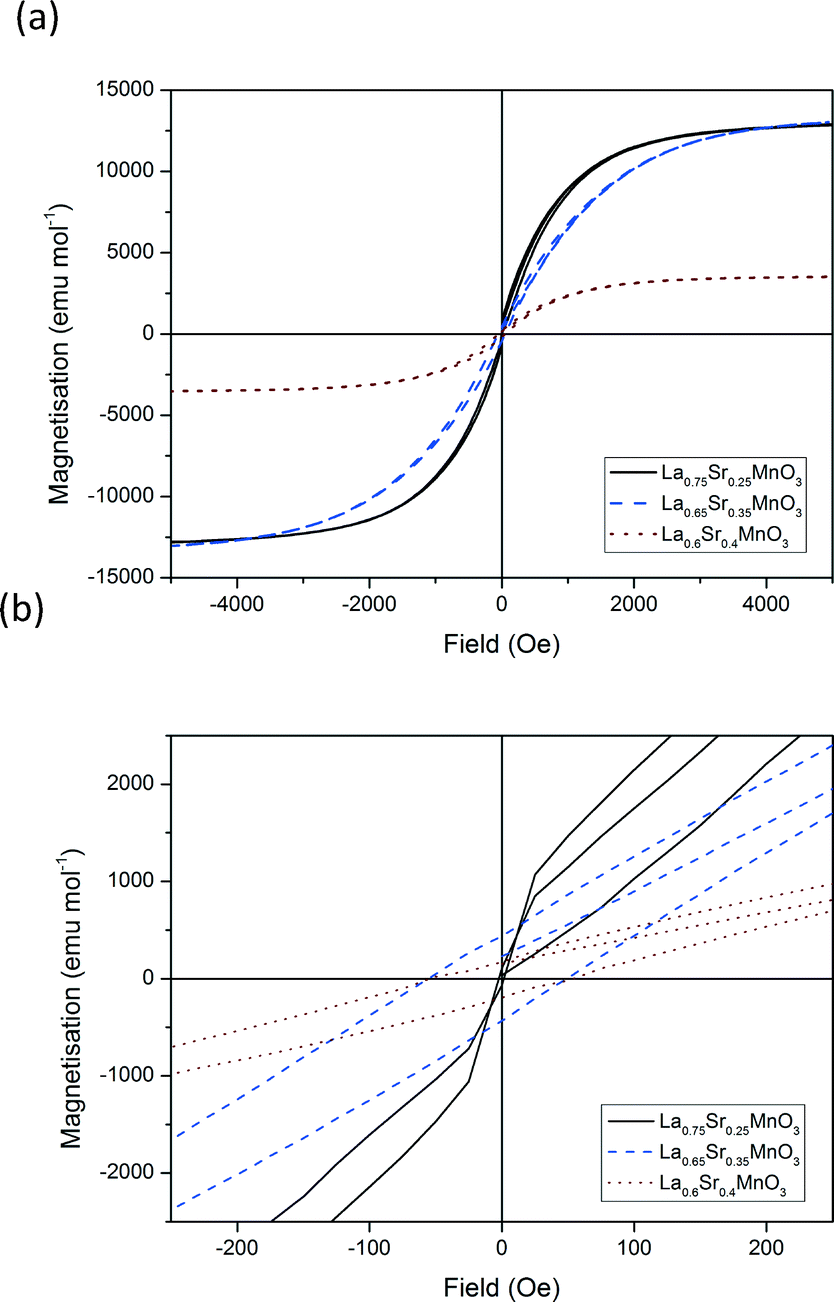
Table 5 Parametric characterisation of hysteresis for a selection of doped LSMO materials (Ms – saturation magnetisation, Hc – coercivity, Br – remanence, W – hysteretic losses and Tc – Curie temperature)
| Sample |
M
s emu mol−1 |
H
c Oe |
B
r emu mol−1 |
W
100k J gMn−1 |
W
150k J gMn−1 |
T
c K |
| LSMO25 |
12800 |
25 |
339 |
5400 |
15900 |
346 |
| LSMO35 |
13000 |
75 |
408 |
4300 |
11400 |
362 |
| LSMO40 |
3500 |
50 |
155 |
1600 |
1000 |
352 |
Fig. 12 shows the hysteresis loops recorded for each of the materials for x = 0.25, 0.35 and 0.4 at 100 K between −5000 and 5000 Oe. The calculated areas of hysteresis are listed in Table 5. These values show a similar trend to that of the susceptibility measurements. Hysteretic losses decrease with increasing dopant from x = 0.25 to x = 0.4. The decrease in hysteretic losses with increasing dopant can be attributed to the increasing substitution of Jahn–Teller active Mn3+ for non Jahn–Teller active Mn4+. The decrease in Jahn–Teller active ions causes a decrease in the extent of the double exchange mechanism and magnetocaloric effect vide priori. Increasing the temperature from 100–150 K increases the hysteretic losses three-fold for LSMO25 and LSMO35. However no significant increase in hysteretic losses is calculated for LSMO40.
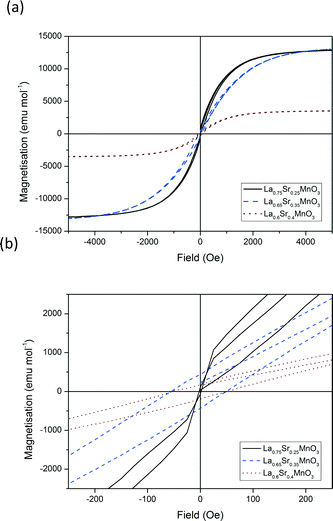 |
| | Fig. 12 Hysteresis loops for a selection of doped LSMO materials at 100 K. (a) Complete field range (−5000 to 5000 Oe) and (b) zoom in from −250 to +250 Oe. | |
An inverse trend is observed for LSMO with respect to hysteretic losses and magnetocaloric effect. It must be stressed however that the two types of measurements (i.e. induction heating and static hysteretic losses) are not directly comparable so no further conclusions can be drawn at this stage.
Conclusions
The polycrystalline structure of a family La1−xSrxMnO3 of perovskite-type MNP clusters with different dopant concentrations was evaluated and linked with the magnetic properties of the materials with the aim to make predictions with respect to the best LSMO composition for use as a mild hyperthermia mediator. Taking into consideration the target temperature range and requirement for a low MNP dosage for therapeutic application, whilst maintaining a high SAR it was found that La0.65Sr0.35MnO3 appeared be the most suitable material. It was found that a transition in the crystalline structure of the perovskite crystal between rhombohedral and orthorhombic increases the magnetocaloric properties of LSMO. FTIR has been shown to be a useful technique in order to compare potential mediators and to predict their magnetocaloric properties. This study presents the magnetic properties (magnetic susceptibility, hysteretic and heating experiment measurements) of the overall sample containing the agglomerated nanoparticles, not the nanoparticles themselves. As we are looking at the magnetic properties of the overall sample as a result of changing the dopant, a TEM study on the nanoparticles would be beyond the scope of this project. Further detailed studies on the stoichiometry of the crystalline phases for the actual magnetic nanoparticles are however planned for future work.
Acknowledgements
Katherine McBride and Suzanne Gray would like to acknowledge financial support from the Department of Education and Learning Northern Ireland (DEL-NI) for a PhD studentship. The authors also acknowledge the help of Professor David Rooney and Christopher McCallum for their help with the induction heating experiments.
Notes and references
- Y. Fukumori and H. Ichikawa, Adv. Powder Technol., 2006, 17, 1–28 CrossRef.
- A. Jordan, R. Scholz, P. Wust, H. Fähling and R. Felix, J. Magn. Magn. Mater., 1999, 201, 413–419 CrossRef CAS.
- L. Asín, M. R. Ibarra, A. Tres and G. F. Goya, Pharm. Res., 2012, 29, 1319–1327, DOI:10.1007/s11095-012-0710-z.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, J. Neuro-Oncol., 2011, 103, 317–324, DOI:10.1007/s11060-010-0389-0.
- F. K. H. van Landeghem, K. Maier-Hauff, A. Jordan, K. Hoffmann, U. Gneveckow, R. Scholz, B. Thiesen, W. Brück and A. von Deimling, Biomaterials, 2009, 30, 52–57 CrossRef CAS PubMed.
- A. Jordan, R. Scholz, P. Wust, H. Schirra, T. Schiestel, H. Schmidt and R. Felix, J. Magn. Magn. Mater., 1999, 194, 185–196 CrossRef CAS.
- O. A. Kuznetsov, O. N. Sorokina, V. G. Leontiev, O. A. Shlyakhtin, A. L. Kovarski and A. A. Kuznetsov, J. Magn. Magn. Mater., 2007, 311, 204–207 CrossRef CAS.
- N. A. Brusentsov, V. V. Gogosov, T. N. Brusentsova, A. V. Sergeev, N. Y. Jurchenko, A. A. Kuznetsov, O. A. Kuznetsov and L. I. Shumakov, J. Magn. Magn. Mater., 2001, 225, 113–117 CrossRef CAS.
- N. A. Brusentsov, T. N. Brusentsova, E. Y. Filinova, N. Y. Jurchenko, D. A. Kupriyanov, Y. A. Pirogov, A. I. Dubina, M. N. Shumskikh, L. I. Shumakov, E. N. Anashkina, A. A. Shevelev and A. A. Uchevatkin, J. Magn. Magn. Mater., 2007, 311, 176–180 CrossRef CAS.
- R. Di Corato, A. Espinosa, L. Lartigue, M. Tharaud, S. Chat, T. Pellegrino, C. Ménager, F. Gazeau and C. Wilhelm, Biomaterials, 2014, 35, 6400–6411 CrossRef CAS PubMed.
- M. Suto, Y. Hirota, H. Mamiya, A. Fujita, R. Kasuya, K. Tohji and B. Jeyadevan, J. Magn. Magn. Mater., 2009, 321, 1493–1496 CrossRef CAS.
- A. Tomitaka, A. Hirukawa, T. Yamada, S. Morishita and Y. Takemura, J. Magn. Magn. Mater., 2009, 321, 1482–1484 CrossRef CAS.
- C. S. S. R. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789–808 CrossRef CAS PubMed.
- T. Sadhukha, T. S. Wiedmann and J. Panyam, Biomaterials, 2014, 35, 7860–7869 CrossRef CAS PubMed.
- P. Pradhan, J. Giri, G. Samanta, H. D. Sarma, K. P. Mishra, J. Bellare, R. Banerjee and D. Bahadur, J. Biomed. Mater. Res., Part B, 2007, 81, 12–22, DOI:10.1002/jbm.b.30630.
- S. Mornet, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161–2175, 10.1039/B402025A.
- S. Mornet, S. Vasseur, F. Grasset, P. Veverka, G. Goglio, A. Demourgues, J. Portier, E. Pollert and E. Duguet, Prog. Solid State Chem., 2006, 34, 237–247 CrossRef CAS.
- A. S. Teja and P. Koh, Prog. Cryst. Growth Charact. Mater., 2009, 55, 22–45 CrossRef CAS.
- I. Apostolova and J. M. Wesselinowa, Solid State Commun., 2009, 149, 986–990 CrossRef CAS.
- A. A. Kuznetsov, V. G. Leontiev, V. A. Brukvin, G. N. Vorozhtsov, B. Y. Kogan, O. A. Shlyakhtin, A. M. Yunin, O. I. Tsybin and O. A. Kuznetsov, J. Magn. Magn. Mater., 2007, 311, 197–203 CrossRef CAS.
- T. L. Kline, Y. Xu, Y. Jing and J. Wang, J. Magn. Magn. Mater., 2009, 321, 1525–1528 CrossRef CAS.
- M. Zeisberger, S. Dutz, R. Müller, R. Hergt, N. Matoussevitch and H. Bönnemann, J. Magn. Magn. Mater., 2007, 311, 224–227 CrossRef CAS.
- D. S. Nikam, S. V. Jadhav, V. M. Khot, M. R. Phadatare and S. H. Pawar, J. Magn. Magn. Mater., 2014, 349, 208–213 CrossRef CAS.
- N. K. Prasad, L. Hardel, E. Duguet and D. Bahadur, J. Magn. Magn. Mater., 2009, 321, 1490–1492 CrossRef CAS.
- E. Pollert, P. Veverka, M. Veverka, O. Kaman, K. Záveta, S. Vasseur, R. Epherre, G. Goglio and E. Duguet, Prog. Solid State Chem., 2009, 37, 1–14 CrossRef CAS.
- S. Vasseur, E. Duguet, J. Portier, G. Goglio, S. Mornet, E. Hadová, K. Knížek, M. Maryško, P. Veverka and E. Pollert, J. Magn. Magn. Mater., 2006, 302, 315–320 CrossRef CAS.
- E. Pollert, K. Knížek, M. Maryško, P. Kašpar, S. Vasseur and E. Duguet, J. Magn. Magn. Mater., 2007, 316, 122–125 CrossRef CAS.
- O. Kaman, P. Veverka, Z. Jirák, M. Maryško, K. Knížek, M. Veverka, P. Kašpar, M. Burian, V. Šepelák and E. Pollert, J. Nanopart. Res., 2011, 13, 1237–1252 CrossRef CAS.
- N. D. Thorat, V. M. Khot, A. B. Salunkhe, R. S. Ningthoujam and S. H. Pawar, Colloids Surf., B, 2013, 104, 40–47 CrossRef CAS PubMed.
- N. D. Thorat, S. V. Otari, R. A. Bohara, H. M. Yadav, V. M. Khot, A. B. Salunkhe, M. R. Phadatare, A. I. Prasad, R. S. Ningthoujam and S. H. Pawar, Mater. Sci. Eng., C, 2014, 42, 637–646 CrossRef CAS PubMed.
- T. K. D. S. Das, J. Phys.: Condens. Matter, 2006, 18, 7629 CrossRef PubMed.
- R. Epherre, C. Pepin, N. Penin, E. Duguet, S. Mornet, E. Pollert and G. Goglio, J. Mater. Chem., 2011, 21, 14990–14998 RSC.
- G. D. V. A. Gaur, J. Phys.: Condens. Matter, 2006, 18, 8837 CrossRef.
- P. Žvátora, M. Veverka, P. Veverka, K. Knížek, E. Pollert, V. Král, G. Goglio, E. Duguet, K. Záveta and O. Kaman, J. Solid State Chem., 2013, 204, 373–379 CrossRef.
- E. Natividad, M. Castro, G. Goglio, I. Andreu, R. Epherre, E. Duguet and A. Mediano, Nanoscale, 2012, 4, 3954–3962, 10.1039/C2NR30667K.
- C. Zener, Phys. Rev., 1951, 82, 403 CrossRef CAS.
-
V. Markovich, A. Wisniewski and H. Szymczak, in Handbook of Magnetic Materials, ed. nonymous, Elsevier, 2014, vol. 22, pp. 1–201 Search PubMed.
- M. Abrashev, V. Ivanov, M. Iliev, R. Chakalov, R. Chakalova and C. Thomsen, Phys. Status Solidi B, 1999, 215, 631–636 CrossRef CAS.
- N. K. Prasad, K. Rathinasamy, D. Panda and D. Bahadur, J. Biomed. Mater. Res., Part B, 2008, 85, 409–416 CrossRef CAS PubMed.
- A. Urushibara, Y. Moritomo, T. Arima, A. Asamitsu, G. Kido and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 14103–14109 CrossRef CAS.
- L. Nalbandian, A. Evdou and V. Zaspalis, Int. J. Hydrogen Energy, 2009, 34, 7162–7172 CrossRef CAS.
-
M. P. Pechini, Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor, US Pat., 3330697 A, 1967 Search PubMed.
- S. Liu, X. Tan, K. Li and R. Hughes, Ceram. Int., 2002, 28, 327–335 CrossRef CAS.
- C. H. Chen, H. Kruidhof, H. J. M. Bouwmeester and A. J. Burggraaf, Mater. Sci. Eng., B, 1996, 39, 129–132 CrossRef.
- R. R. Wildeboer, P. Southern and Q. A. Pankhurst, J. Phys. D: Appl. Phys., 2014, 47, 495003 CrossRef.
- S. Anisimov, B. Kapeliovich and T. Perelman, Zh. Eksp. Teor. Fiz., 1974, 66, 375–377 Search PubMed.
- A. Da Silva, L. D. Conceição, A. Rocco and M. Souza, Ceramica, 2012, 58, 521–528 CAS.
- N. Zhang, W. Yang, W. Ding, D. Xing and Y. Du, Solid State Commun., 1999, 109, 537–542 CrossRef CAS.
-
M. Dokiya, Proceedings of the Fourth International Symposium on Solid Oxide Fuel Cells (SOFC-IV), The Electrochemical Society, 1995 Search PubMed.
-
D. Green and K. Neumann, arXiv preprint arXiv:1105.2700, 2011 Search PubMed.
- A. Mellergård, R. McGreevy and S. Eriksson, J. Phys.: Condens. Matter, 2000, 12, 4975 CrossRef.
- A. Rostamnejadi, H. Salamati, P. Kameli and H. Ahmadvand, J. Magn. Magn. Mater., 2009, 321, 3126–3131 CrossRef CAS.
- Z. F. Zi, Y. P. Sun, X. B. Zhu, Z. R. Yang, J. M. Dai and W. H. Song, J. Magn. Magn. Mater., 2009, 321, 2378–2381 CrossRef CAS.
- R. T. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction data, elemental compositions determined by EDX. See DOI: 10.1039/c5ce01890k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
J.
Cook
ab,
S.
Gray
b,
S.
Felton
b,
L.
Stella
ab and
D.
Poulidi
*a
a,
J.
Cook
ab,
S.
Gray
b,
S.
Felton
b,
L.
Stella
ab and
D.
Poulidi
*a