
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and crystal structures of twisted three-dimensional assemblies of adamantane-bridged tris-NHC ligands and AgI†
Masahide
Tominaga
*a,
Taichi
Kawaguchi
a,
Kazuaki
Ohara
a,
Kentaro
Yamaguchi
a,
Hyuma
Masu
b and
Isao
Azumaya
*c
aFaculty of Pharmaceutical Sciences at Kagawa Campus, Tokushima Bunri University, 1314-1 Shido, Sanuki, Kagawa 769-2193, Japan. E-mail: tominagam@kph.bunri-u.ac.jp
bCenter for Analytical Instrumentation, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
cFaculty of Pharmaceutical Sciences, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan. E-mail: isao.azumaya@phar.toho-u.ac.jp
First published on 8th December 2015
Abstract
Two adamantane-bridged tripodal trisimidazolium or trisbenzimidazolium salts were designed as C3v-symmetrical tricarbene ligands and synthesized in good yields. The reaction of the tripodal trisazolium salt precursors with silver oxide provided trisilver hexacarbene complexes bearing a three-dimensional framework. Multinuclear NMR spectroscopy and mass spectrometry data showed that the complexes were symmetric structures of two tricarbene ligands, three metal ions, and three counter anions. X-ray crystallographic analyses revealed that both complex cations adopted a highly twisted conformation. The complex cations built from the trisimidazolium salts were arranged into columnar structures through CH/π interactions, which were assembled into a three-dimensional network structure by intermolecular CH/F interactions via hexafluorophosphate anions. The complex cations generated from the trisbenzimidazolium salts were aligned into columnar structures, which were arranged into a network structure through CH/π interactions. Solvents and counter anions were accommodated in the spaces of the crystalline lattices.
Introduction
Metal-mediated three-dimensional (3D) structures have gained attention because of their potential applications in molecular encapsulation, cavity-directed catalytic transformations, and stabilization of reactive intermediates.1–4 Various types of 3D structures including cages, capsules, cylinders, boxes, and tubes have been made from appropriate combinations of multidentate organic ligands and metal ions. These metal–organic frameworks based on Werner-type metal complexes have been of particular interest for the last two decades. Recently, many different N-heterocyclic carbenes (NHCs) and their metal complexes have received interest owing to a wide variety of applications in homogeneous and asymmetric catalysis and luminescence.5–7 The production of discrete 3D metallosupramolecular complexes in cylinder-like and tubular structures from organometallic frameworks has been successfully realized through the formation of stable M–CNHC bonds between poly-NHC ligands and metal atoms.8 For instance, Meyer and co-workers have constructed a silver NHC complex from a neopentane-based tripodal carbene ligand.9 Hahn and co-workers showed that a series of disk-shaped poly-NHC ligands could self-assemble into multinuclear cylinder-like structures of various sizes and shapes.10 The preparation of box- and cylinder-like structures from macrocyclic poly-NHC ligands has been reported by several groups.11 The design of these poly-NHC ligands is crucial in determining the size, shape, and conformation of the resulting organometallic assembly. C3-symmetric, relatively flexible C3v-symmetrical and macrocyclic NHC ligands have been used; however, the use of rigid C3v-symmetrical tricarbene ligands for the creation of well-defined complexes remains less explored. Herein, we report the efficient preparation of two adamantane-derived tris-NHC ligands bearing imidazolium or benzimidazolium salts and their trisilver hexacarbene complexes. X-ray crystallographic analysis revealed that both complexes possess a 3D organometallic framework that has an exceedingly twisted conformation in the solid state. The helical trisilver complex cations were arranged into columnar structures, which further assembled into network structures by multiple intermolecular interactions.We have previously shown that adamantane derivatives can be useful building blocks for the design and synthesis of macrocycles and cages, and allow unique guest-inclusion abilities and self-assembly behaviors over the past few years.12 We selected trisubstituted adamantane derivatives for further investigation, as they have been versatile candidates for C3v-symmetric tricarbene ligands. They are rigid and well-defined, and have highly symmetric skeletons. Thus, we have designed novel molecular-based organometallic assemblies, which were constructed from two tripodal 1,3,5-triphenyladamantane-derived trisimidazolium or trisbenzimidazolium salts and three metal ions.
Results and discussion
The synthesis of the trisimidazolium salt H3-2(PF6)3 is depicted in Scheme 1. The copper(I)-catalyzed reaction of imidazole with 1,3,5-tris(4-iodophenyl)adamantane gave the corresponding tripodal adamantane derivative (1). Alkylation of the three imidazoles in 1 with ethyl bromide gave the product after anion exchange with ammonium hexafluorophosphate, H3-2(PF6)3. The reaction of silver oxide (0.15 mmol) with the trisimidazolium salt H3-2(PF6)3 (0.10 mmol) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry in acetonitrile (10 mL) at 50 °C for 2 days resulted in the formation of the trisilver hexacarbene complex [Ag3(2)2](PF6)3 (3) in 81% yield. The 1H NMR spectrum of the product showed that the peak corresponding to the imidazolium proton in H3-2(PF6)3 at 8.91 ppm had disappeared, inferring the production of a carbene complex. In the 13C NMR spectrum of 3, the resonance from the carbene carbon atoms in the complex was observed at 180.0 ppm, which is consistent with values previously reported for other silver carbene complexes.10 Furthermore, only four signals are observed in the aromatic region of the 1H NMR spectrum of 3, indicating that the ligands are equivalent and symmetric in the product. In addition to the NMR analysis, cold-spray ionization MS (CSI-MS) confirmed the desired composition of 3, having three silver atoms, three hexafluorophosphate anions, and two adamantane-bridged imidazolin-2-ylidene ligands. Two peaks were found in the mass spectrum: at m/z 1907.4020, corresponding to [M − PF6]+, and m/z 881.2183, corresponding to [M − 2PF6]2+ (Fig. S1 in the ESI†). The isotopic distributions in the peaks were consistent with their calculated theoretical pattern. Complex 3 can be dissolved in many organic solvents including dimethyl sulfoxide, methanol, acetonitrile, and acetone, but is only sparingly soluble in chloroform, diethyl ether, hexane, and toluene.
:2 stoichiometry in acetonitrile (10 mL) at 50 °C for 2 days resulted in the formation of the trisilver hexacarbene complex [Ag3(2)2](PF6)3 (3) in 81% yield. The 1H NMR spectrum of the product showed that the peak corresponding to the imidazolium proton in H3-2(PF6)3 at 8.91 ppm had disappeared, inferring the production of a carbene complex. In the 13C NMR spectrum of 3, the resonance from the carbene carbon atoms in the complex was observed at 180.0 ppm, which is consistent with values previously reported for other silver carbene complexes.10 Furthermore, only four signals are observed in the aromatic region of the 1H NMR spectrum of 3, indicating that the ligands are equivalent and symmetric in the product. In addition to the NMR analysis, cold-spray ionization MS (CSI-MS) confirmed the desired composition of 3, having three silver atoms, three hexafluorophosphate anions, and two adamantane-bridged imidazolin-2-ylidene ligands. Two peaks were found in the mass spectrum: at m/z 1907.4020, corresponding to [M − PF6]+, and m/z 881.2183, corresponding to [M − 2PF6]2+ (Fig. S1 in the ESI†). The isotopic distributions in the peaks were consistent with their calculated theoretical pattern. Complex 3 can be dissolved in many organic solvents including dimethyl sulfoxide, methanol, acetonitrile, and acetone, but is only sparingly soluble in chloroform, diethyl ether, hexane, and toluene.
 | ||
| Scheme 1 Preparation of the trisimidazolium salt H3-2(PF6)3 and the complex [Ag3(2)2](PF6)3 (3). | ||
The molecular structure of complex 3 was determined by single-crystal X-ray analysis (Fig. 1). Single crystals were obtained from vapor diffusion of 1,4-dioxane into an acetonitrile solution of 3. The compound crystallized in the space group P21/c, which contains one molecule of 3 and six molecules of 1,4-dioxane in the asymmetric unit. The molecular structure of the complex cation [Ag3(2)2]3+ showed a 3D structure with a twisted conformation. The complex had small internal spaces within the framework, which were too small to allow for the encapsulation of small organic molecules. The centroid–centroid distance between the two adamantane moieties is 6.66 Å, and the three distances between the silver atoms are 12.53, 12.92, and 13.36 Å. The complex cation has chirality, arising from the direction of the twist in the triphenyladamantane moieties, where the torsion angle between the two triphenyladamantane parts of 3 is 43.0°. There exist two pairs of enantiomeric conformers of the complex cation with (M)- and (P)-helicity in the unit cell, meaning that the crystals have no overall chirality. The three pairs of phenyl rings are nearly vertical to each other, where the angles between the planes of the benzene moieties are 65.3, 76.2, and 87.3°. The centers of the three pairs of phenyl rings are separated by 4.99–5.01 Å. The torsion angles between the planes of the phenyl and imidazolin-2-ylidene rings range from 35.6 to 50.5°. The distances of Ag–Ccarbene and the Ccarbene–Ag–Ccarbene angles in 3 are 2.06–2.09 Å and 174.8–178.8°, respectively. It is noteworthy that the angles between the plane of the three metal centers and the plane of the metal-mediated imidazolin-2-ylidene rings are 12.0, 14.5, and 20.6°. Each complex cation has a columnar structure formed through CH/π interactions between the adamantane parts and imidazolin-2-ylidene rings and phenyl groups along the c axis (Fig. 2a), containing alternating enantiomers of the complex cation. These columns interacted with the hexafluorophosphate anions via multiple CH/F interactions13 between the hydrogen atoms of the imidazolin-2-ylidene, ethyl, and phenyl groups and the fluorine atoms of the hexafluorophosphate anions (Fig. 2b). This gives an overall 3D network. The solvent molecules were accommodated within the spaces of the network structures.
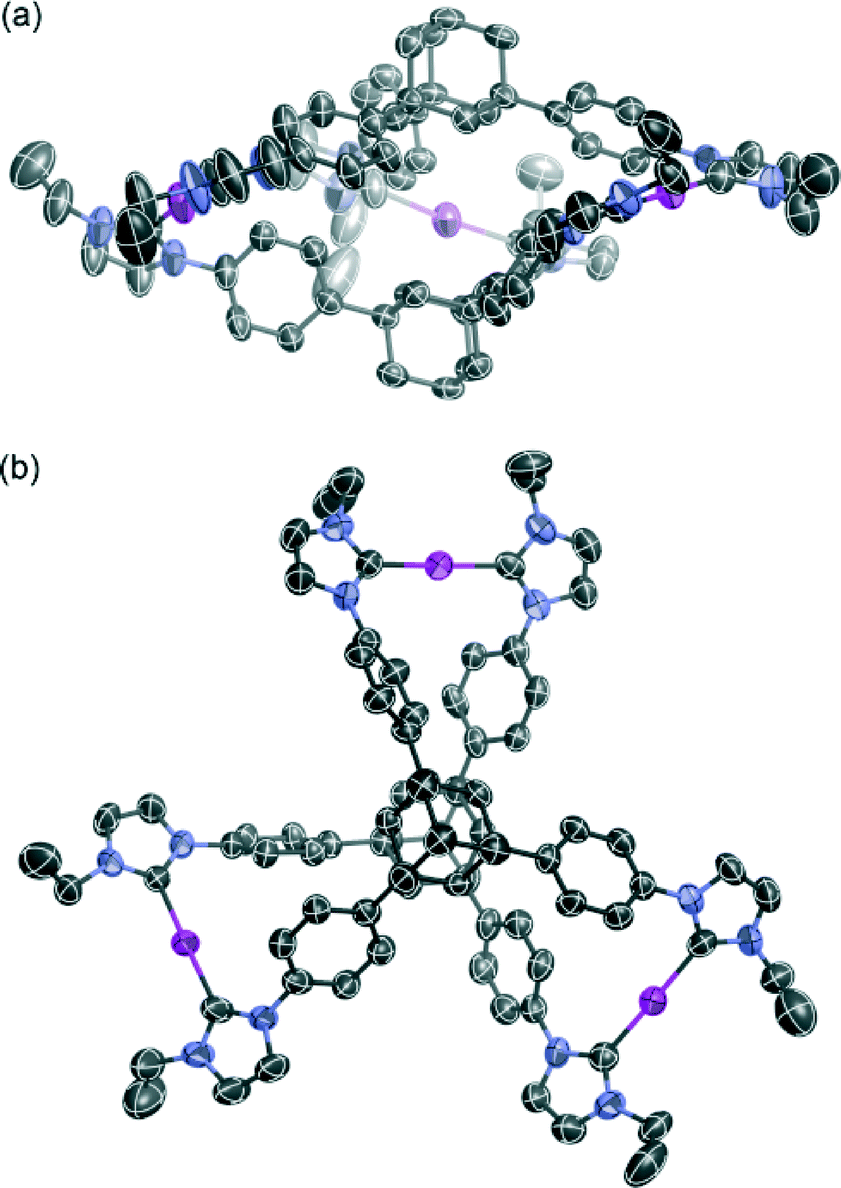 | ||
| Fig. 1 The molecular structure of the complex cation in 3. (a) Side view and (b) top view. Ellipsoids are drawn at the 50% probability level. Solvent molecules, counter anions, and hydrogen atoms have been omitted for clarity. | ||
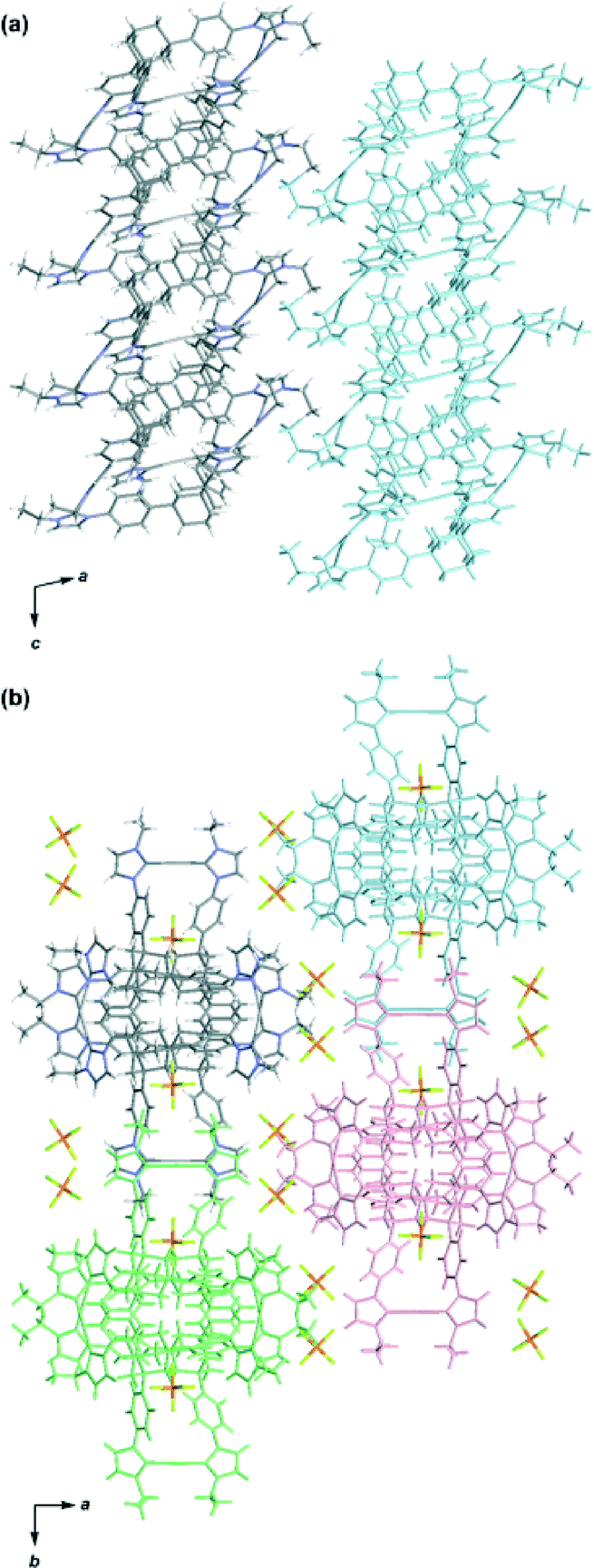 | ||
| Fig. 2 Packing diagram of the complex [Ag3(2)2](PF6)3 (3). (a) Side view of the columns parallel to the c axis; included solvent molecules and counter anions were omitted for clarity. (b) Top view of the network structure; solvent molecules were omitted for clarity. The molecules of neighbouring columns are colored light blue, pink, and light green. | ||
Similarly, we constructed a trisilver hexacarbene complex [Ag3(5)2](PF6)3 (6), where an adamantane-derived trisbenzimidazolium salt was used as the tris-NHC ligand. The trisbenzimidazolium salt H3-5(PF6)3 was synthesized in good yield according to Scheme 2. Mixing silver oxide (0.15 mmol) and H3-5(PF6)3 (0.10 mmol) in a 3:2 stoichiometry gave the trisilver hexacarbene complex [Ag3(5)2](PF6)3 (6) in 85% yield. 1H and 13C NMR data for the product indicated similar results to the complex [Ag3(2)2](PF6)3 (3). The mass spectrum had two peaks: at m/z 1031.2670, corresponding to [M − 2PF6]2+, and m/z 639.1885, corresponding to [M − 3PF6]3+, indicating that it was the desired complex (Fig. S2 in the ESI†). Complex 6 dissolves in many organic solvents including dimethyl sulfoxide, acetonitrile, and acetone, but is only sparingly soluble in methanol, hexane, chloroform, diethyl ether, and toluene.
 | ||
| Scheme 2 Preparation of the trisbenzimidazolium salt H3-5(PF6)3 and the complex [Ag3(5)2](PF6)3 (6). | ||
Single crystals were obtained from the vapor diffusion of 1,4-dioxane into an acetonitrile solution of 6. The complex crystallized in the space group P21/c, and has one molecule of 6, two molecules of 1,4-dioxane and one molecule of acetonitrile in the asymmetric unit. In the crystal structure of 6, the complex cation [Ag3(5)2]3+ adopted a similar conformation to [Ag3(2)2]3+ despite the introduction of the bulky benzimidazolin-2-ylidene ring (Fig. 3). The centroid–centroid distance between the two adamantane moieties is 6.69 Å, and the three distances between the silver atoms are 12.46, 12.87, and 12.89 Å. The torsion angle between the two triphenyladamantane parts of 6 is 43.3°. Again, the complex cation contains two helical enantiomers per unit cell. In the three pairs of phenyl rings, the two pairs are nearly vertical, and the other is almost parallel to each other. The angles between the ring planes of the benzene moieties are 7.5, 63.5, and 78.2°. The distances between the centers of the three pairs of phenyl rings are 4.83, 5.13, and 5.25 Å. The torsion angles between the planes of the phenyl and benzimidazolin-2-ylidene rings range from 48.4 to 55.7°. The distances of Ag–Ccarbene and the angles of Ccarbene–Ag–Ccarbene in 6 are 2.07–2.10 Å and 175.2–177.9°. The angles between the plane of the three metal centers and the plane of the metal-mediated benzimidazolin-2-ylidene rings are 8.7, 19.4, and 22.5°. Individual complex cations were arranged into a columnar structure via CH/π interactions between benzimidazolin-2-ylidene, phenyl, and adamantyl groups along the b axis (Fig. 4a), containing alternating enantiomers of the complex cation. The columns were assembled into a network structure through CH/π interactions between benzimidazolin-2-ylidene, phenyl, and ethyl groups (Fig. 4b). Hexafluorophosphate anions interacted with benzimidazolin-2-ylidene, phenyl, ethyl, and adamantyl groups via CH/F interactions. Solvent molecules were included in the spaces of the network structures.
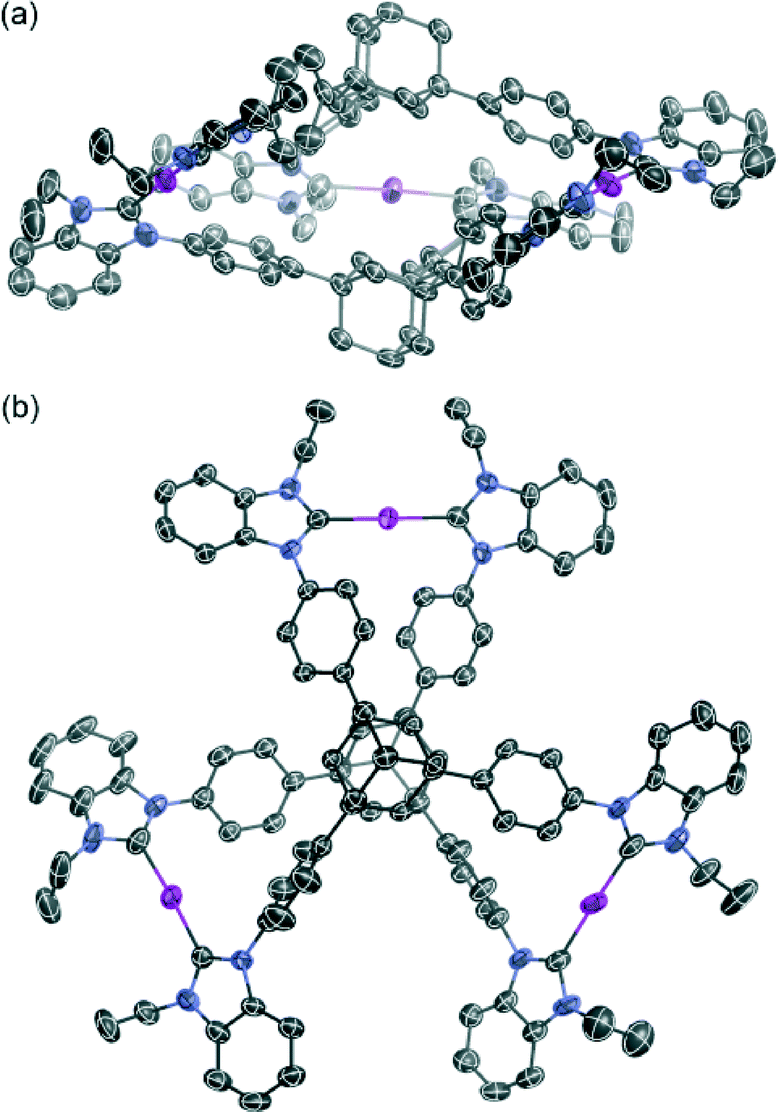 | ||
| Fig. 3 The molecular structure of the complex cation in 6. (a) Side view and (b) top view. Ellipsoids are drawn at the 50% probability level. Solvent molecules, counter anions, disordered atoms, and hydrogen atoms have been omitted for clarity. | ||
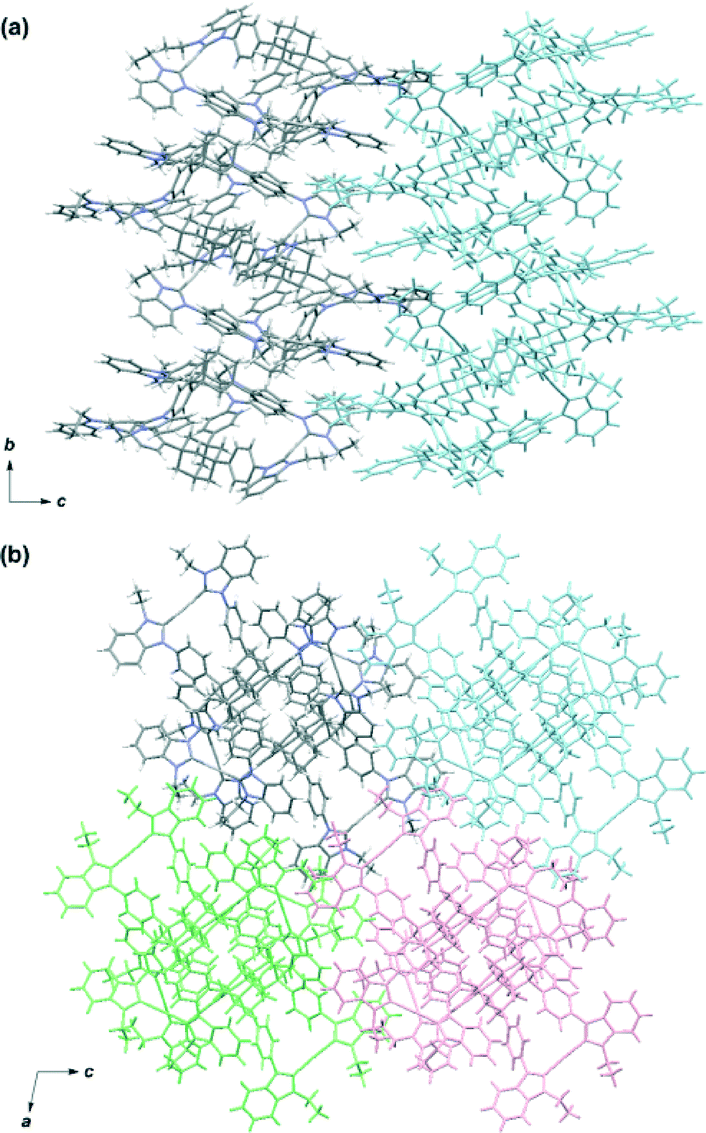 | ||
| Fig. 4 Packing diagram of the silver complex [Ag3(5)2](PF6)3 (6). (a) Side view of the columnar structure along the b axis. (b) Top view of the network structure. Solvent molecules and counter anions were omitted for clarity. The molecules of neighbouring columns are colored light blue, pink, and light green. | ||
Hahn and Peris et al. demonstrated that C3-symmetrical tricarbene ligands can form trinuclear silver(I), gold(I), and copper(I) carbene complexes, which adopt a twisted conformation in the solid state.10 In comparison with these complex cations, the distortions between the rigid tripodal parts in 3 and 6 are similar. The angles between the plane of the three metal centers and the plane of the metal-mediated imidazolin-2-ylidene or benzimidazolin-2-ylidene rings were nearly parallel, showing the helical structures having enormously twisted conformations. Recently, a double-stranded NHC-helicate from a pentadentate diNHC ligand was studied by Huynh and co-workers.14a The research on extended organometallic frameworks containing this twisted complex cation is quite restricted,14 whereas there are several examples of twisted structures made up of Werner-type metal–organic frameworks.15 Accordingly, tripodal adamantane-based tricarbene ligands of non-disk-shaped C3v-symmetrical molecules to bend disk-shaped C3-symmetrical compounds are applicable building blocks for the creation of helical organometallic assemblies.
Conclusions
In summary, we have demonstrated that tripodal adamantane-derived tris-NHC ligands can be used for the construction of well-defined 3D trisilver hexacarbene complexes with a twisted conformation. This was achieved through metal-directed self-assembly, where two tricarbene ligands were bridged with three metal atoms. Two metallosupramolecular structures were obtained in high yields without the generation of other oligomeric and polymeric by-products, and this shows that the use of non-disk-shaped C3v-symmetrical ligands does not inhibit the self-assembly process. These results provide a strategy for the design of twisted 3D organometallic assemblies and the exploitation of helical structures. Studies on the preparation and applications of 3D NHC complexes bearing other metal centers and chiral counter anions are ongoing in our laboratory.Experimental
General procedure
All reagents and solvents were obtained from commercial suppliers and used without further purification. The synthesis of 1,3,5-tris(4-iodophenyl)adamantane was performed according to the literature.16 Melting points were determined using an ATM-01. IR spectra were recorded on a Jasco FT/IR-6300. 1H and 13C NMR spectra were recorded on a Bruker AV400 spectrometer at 298 K in CDCl3 or DMSO-d6 with trimethylsilane as the reference, and CD3OD or CD3CN. X-ray crystal structure data were collected using a Bruker SMART APEX II diffractometer with Mo Kα radiation and a Bruker D8 VENTURE diffractometer with Cu Kα radiation. Column chromatography was performed on a Wakogel C200, and thin-layer chromatography was carried out on 2.0 mm Merck precoated silica gel glass plates. Gel permeation chromatography was performed using a recycling preparative HPLC (LC-9204, Japan Analytical Industry Co., Ltd.) and a JAIGEL H series column (Japan Analytical Industry Co., Ltd.). HRMS and CSI-MS measurements were performed using a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS; Apex-Qe 9.4 T, Bruker Daltonics, Inc. Billerica, MA). We could not perform satisfactory elemental analyses of the precursors and the complexes in several attempts, presumably due to their hydroscopic nature and the large amount of fluorine present, and to the capability of their molecules to keep the solvents.Synthesis of 1,3,5-tris(4-(imidazol-1-yl)phenyl)adamantane (1)
A mixture of 1,3,5-tris(4-iodophenyl)adamantane (7.42 g, 10.0 mmol), potassium carbonate (8.29 g, 60.0 mmol), N,N-dimethylglycine (0.62 g, 6.0 mmol), imidazole (4.08 g, 60.0 mmol), and CuI (0.57 g, 3.0 mmol) in dry dimethyl sulfoxide (100 mL) was stirred at 120 °C for 4 days under an argon atmosphere. The reaction mixture was evaporated under reduced pressure. The residue was dissolved in chloroform and water, and the solution was washed with water and brine and dried over sodium sulfate. After removal of the solvent, silica gel column chromatography (eluent: CHCl3:MeOH = 20:1) and gel permeation chromatography (eluent: CHCl3) of the residue afforded the title compound as a white solid (4.20 g, 7.46 mmol) in 75% yield. M.p. 162–163 °C. FT-IR (ATR, cm−1): 2916, 2851, 1521, 1507, 1490, 1304, 1219, 1054, 964, 772, 728. 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 3H), 7.56 (d, J = 8.8 Hz, 6H), 7.39 (d, J = 8.4 Hz, 6H), 7.28 (s, 3H), 7.21 (s, 3H), 2.65 (br s, 1H), 2.20–2.13 (m, 6H), 2.08 (br d, J = 2.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 148.9, 135.6, 135.5, 130.4, 126.4, 121.5, 118.3, 48.0, 41.2, 38.3, 29.9. HRMS (ESI, m/z) calcd for C37H35N6 [M + H]+ 563.2918, found 563.2912.
Synthesis of 1,3,5-tris(4-(3-ethylimidazol-1-yl)phenyl)adamantane tribromide, H3-2(Br)3
1,3,5-Tris(4-(imidazol-1-yl)phenyl)adamantane (1) (0.42 g, 0.75 mmol) was dissolved in dry N,N-dimethylformamide (20 mL), and ethyl bromide (0.98 g, 9.0 mmol) was added. The reaction mixture was stirred at 50 °C for 2 days under an argon atmosphere. The reaction mixture was evaporated under reduced pressure. The white solids were dissolved in methanol, and diethyl ether was added. The resulting precipitate was isolated by filtration and, after washing with diethyl ether, the title compound was dried in vacuo to give a white solid (0.53 g, 0.60 mmol) in 79% yield. M.p. 244–246 °C. FT-IR (ATR, cm−1): 2922, 2850, 1653, 1550, 1516, 1457, 1260, 1210, 1140, 1071, 1018, 828, 771, 746. 1H NMR (400 MHz, CD3OD) δ 9.67 (s, 3H), 8.11 (s, 3H), 7.92 (s, 3H), 7.84–7.78 (m, 12H), 4.44 (q, J = 7.2 Hz, 6H), 2.62 (br s, 1H), 2.29–2.11 (m, 12H), 1.65 (t, J = 7.2 Hz, 9H). 13C NMR (100 MHz, CD3OD) δ 153.4, 136.0, 134.3, 128.4, 124.2, 123.1, 122.8, 48.6, 46.6, 41.9, 39.8, 31.4, 15.6. HRMS (ESI, m/z) calcd for C43H47N6 [M − 2H − 3Br]+ 647.3857, found 647.3855.Synthesis of 1,3,5-tris(4-(3-ethylimidazol-1-yl)phenyl)adamantane tris(hexafluorophosphate), H3-2(PF6)3
A solution of ammonium hexafluorophosphate (0.38 g, 2.3 mmol) in methanol (3.0 mL) was added to a methanol solution (7.0 mL) of compound H3-2(Br)3 (0.53 g, 0.60 mmol). The white hexafluorophosphate salt precipitated instantly. Water was added into the reaction mixture and stirred for 12 h at room temperature. The precipitates were isolated by filtration and, after washing with methanol and diethyl ether, the title compound was dried in vacuo to give a white solid (0.62 g, 0.57 mmol) in 95% yield. M.p. 266–267 °C. FT-IR (ATR, cm−1): 2929, 2858, 1653, 1559, 1516, 1507, 1457, 1213, 1078, 1021, 838, 772, 739. 1H NMR (400 MHz, CD3CN) δ 8.91 (s, 3H), 7.80 (d, J = 8.8 Hz, 6H), 7.78 (t, J = 2.0 Hz, 3H), 7.63–7.60 (d, J = 8.8 Hz and s, 9H), 4.29 (q, J = 7.2 Hz, 6H), 2.60 (br t, J = 2.8 Hz, 1H), 2.25–2.17 (m, 6H), 2.09 (br d, J = 2.4 Hz, 6H), 1.56 (t, J = 7.2 Hz, 9H). 13C NMR (100 MHz, CD3CN) δ 153.3, 135.2, 133.9, 128.3, 123.9, 123.2, 122.7, 47.8, 46.4, 41.5, 39.6, 31.1, 15.3. HRMS (ESI, m/z) calcd for C43H49F12N6P2 [M − PF6]+ 939.3297, found 939.3252.Synthesis of [Ag3(2)2](PF6)3 (3)
Compound H3-2(PF6)3 (0.11 g, 0.10 mmol) was dissolved in dry acetonitrile (10 mL), and silver oxide (35 mg, 0.15 mmol) was added. The suspension was stirred at 50 °C for 2 days under exclusion of light and an argon atmosphere. After cooling to room temperature, the resulting mixture was filtered over celite to give a solution. The filtrate was concentrated to 2 mL, and the addition of diethyl ether into this solution resulted in the precipitation of a white solid. The precipitate was isolated by filtration upon washing with diethyl ether, and the title compound was dried in vacuo to give a white solid (0.08 g, 0.04 mmol) in 81% yield. M.p. > 300 °C (decomposed). FT-IR (ATR, cm−1): 2921, 2852, 1559, 1516, 1471, 1424, 1267, 1223, 1017, 951, 837, 736. 1H NMR (400 MHz, CD3CN) δ 7.55 (d, J = 8.4 Hz, 12H), 7.52 (d, J = 2.0 Hz, 6H), 7.43 (d, J = 1.6 Hz, 6H), 7.27 (d, J = 8.4 Hz, 12H), 4.30 (q, J = 6.4 Hz, 12H), 2.47 (br s, 2H), 2.06 (br d, J = 12.0 Hz, 6H), 1.99 (br s, 12H), 1.89 (br d, J = 12.0 Hz, 6H), 1.53 (t, J = 7.2 Hz, 18H). 13C NMR (100 MHz, CD3CN) δ 180.0, 150.9, 139.2, 127.2, 124.6, 123.1, 122.8, 49.0, 48.2, 41.3, 39.2, 30.7, 17.4. HRMS (CSI, m/z) calcd for C86H92Ag3F6N12P [M − 2PF6]2+ 881.2175, found 881.2183.Synthesis of 1,3,5-tris(4-(benzimidazol-1-yl)phenyl)adamantane (4)
The title compound was synthesized in 84% yield as a white solid in a similar manner to the preparation of 1,3,5-tris(4-(imidazol-1-yl)phenyl)adamantane (1). M.p. 168–169 °C. FT-IR (ATR, cm−1): 2922, 2854, 1516, 1489, 1455, 1340, 1284, 1229, 1048, 976, 839, 738. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 3H), 7.90 (d, J = 5.2 Hz, 3H), 7.70 (d, J = 8.8 Hz, 6H), 7.57–7.52 (m, 9H), 7.37–7.32 (m, 6H), 2.73 (br s, 1H), 2.32–2.24 (m, 6H), 2.17 (br d, J = 2.4 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 149.5, 144.1, 142.3, 134.5, 134.0, 126.7, 124.0, 123.7, 122.8, 120.7, 110.5, 48.1, 41.2, 38.5, 30.0. HRMS (ESI, m/z) calcd for C49H41N6 [M + H]+ 713.3387, found 713.3381.Synthesis of 1,3,5-tris(4-(3-ethylbenzimidazol-1-yl)phenyl)adamantane tribromide, H3-5(Br)3
The title compound was synthesized in 72% yield as a white solid in a similar manner to the preparation of H3-2(Br)3. M.p. 243–245 °C. FT-IR (ATR, cm−1): 2920, 2854, 1559, 1540, 1507, 1472, 1340, 1233, 1191, 1041, 1013, 986, 839, 779. 1H NMR (400 MHz, CD3OD) δ 9.93 (s, 3H), 8.12 (d, J = 7.6 Hz, 3H), 7.99 (d, J = 8.8 Hz, 6H), 7.87–7.74 (m, 15H), 4.71 (q, J = 7.2, Hz, 6H), 2.73 (br s, 1H), 2.46–2.24 (m, 12H), 1.76 (t, J = 7.2 Hz, 9H). 13C NMR (100 MHz, CD3OD) δ 154.2, 142.6, 133.3, 132.9, 132.7, 128.9, 128.7, 128.6, 126.2, 114.8, 114.7, 44.18, 44.16, 42.0, 40.1, 31.6, 14.6. HRMS (ESI, m/z) calcd for C55H54BrN6 [M − H − 2Br]+ 877.3588, found 877.3585.Synthesis of 1,3,5-tris(4-(3-ethylbenzimidazol-1-yl)phenyl)adamantane tris(hexafluorophosphate), H3-5(PF6)3
The title compound was synthesized in 81% yield as a white solid in a similar manner to the preparation of H3-2(PF6)3. M.p. > 300 °C (decomposed). FT-IR (ATR, cm−1): 2922, 2855, 1653, 1559, 1540, 1507, 1472, 1340, 1272, 1231, 1190, 1041, 986, 840, 746. 1H NMR (400 MHz, CD3CN) δ 9.23 (s, 3H), 8.01 (d, J = 8.4 Hz, 3H), 7.94 (d, J = 8.4 Hz, 6H), 7.80–7.72 (m, 15H), 4.60 (q, J = 7.2 Hz, 6H), 2.70 (br s, 1H), 2.39–2.29 (m, 6H), 2.20 (br s, 6H), 1.69 (t, J = 7.6 Hz, 9H). 13C NMR (100 MHz, CD3CN) δ 153.9, 141.5, 133.0, 132.5, 132.2, 128.8, 128.5, 128.4, 126.0, 114.70, 114.68, 48.0, 44.1, 41.5, 39.8, 31.1, 14.5. HRMS (ESI, m/z) calcd for C55H55F12N6P2 [M − PF6]+ 1089.3766, found 1089.3756.Synthesis of [Ag3(5)2](PF6)3 (6)
The title compound was synthesized in 85% yield as a white solid in a similar manner to the preparation of [Ag3(2)2](PF6)3 (3). M.p. > 300 °C (decomposed). FT-IR (ATR, cm−1): 2922, 2855, 1654, 1559, 1516, 1507, 1472, 1342, 1271, 1229, 1190, 1013, 839, 746. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 8.4 Hz, 6H), 7.74 (d, J = 8.4 Hz, 12H), 7.65–7.61 (m, 6H), 7.54–7.48 (m, 12H), 7.44 (d, J = 8.0 Hz, 12H), 4.75 (br d, J = 6.0 Hz, 12H), 2.15 (br d, J = 11.2 Hz, 6H), 2.03–2.00 (m, 18H), 1.63 (t, J = 7.2 Hz, 18H). 13C NMR (100 MHz, DMSO-d6) δ 188.2, 150.0, 135.7, 133.2, 133.0, 126.3, 125.2, 124.8, 112.5, 112.0, 47.9, 44.2, 38.0, 29.2, 16.0. HRMS (CSI, m/z) calcd for C110H104Ag3N12 [M − 3PF6]3+ 639.1881, found 639.1885.X-ray crystallography
X-ray data for crystal 3 were collected on a CCD diffractometer with monochromated Mo Kα (λ = 0.71073 Å) radiation. Data collection was carried out at 150 K using a Japan Thermal Eng. Col., Ltd. Cryostat system equipped with a liquid nitrogen generator. X-ray data for crystal 6 were collected on a CMOS diffractometer (PHOTON 100) with monochromated Cu Kα (λ = 1.54178 Å) radiation. Data collection was carried out at 100 K using a Japan Thermal Eng. Col., Ltd. Cryostat system equipped with a liquid nitrogen generator. The crystal structures of 3 and 6 were solved by direct methods using SHELXS 97 and SHELXTL-2013, respectively.17 Refinements were carried out by full-matrix least squares (on F2) with anisotropic temperature factors for non-H atoms. In all of the structures, H atoms were included in their calculated positions. For refinement of the structure and structure analysis, the program package SHELXTL was used. Treatments of included disordered solvent molecules were executed using the program PLATON.18[Ag3(2)2](PF6)3 (3)
C86H92Ag3N12·3(PF6)·6(C4H8O2), Mr = 2580.85, 0.4 × 0.4 × 0.2 mm3, monoclinic, P21/c, a = 34.813(3), b = 20.6402(19), c = 16.3040(15) Å, β = 102.6780(10)°, V = 11429.6(18) Å3, Z = 4, Dc = 1.500 Mg m−3, 2θmax = 52.93°, T = 150 K, 55296 reflections measured, 22635 unique (Rint = 0.0442), μ = 0.644 mm−1, Tmax = 0.88, Tmin = 0.78. The final R1 and wR2(F2) were 0.0707 and 0.1916 (I > 2σ(I)), 0.1336 and 0.2158 (all data) for 1340 parameters and 96 restraints. CCDC-1420798.
[Ag3(5)2](PF6)3 (6)
C110H104Ag3N12·3(PF6)·2(C4H8O2)·C2N, Mr = 2566.82, 0.27 × 0.18 × 0.08 mm3, monoclinic, P21/c, a = 19.0224(7), b = 15.7764(7), c = 38.4287(15) Å, β = 102.187(2)°, V = 11272.7(8) Å3, Z = 4, Dc = 1.512 Mg m−3, 2θmax = 130.53°, T = 100 K, 101361 reflections measured, 19114 unique (Rint = 0.0593), μ = 5.306 mm−1, Tmax = 0.68, Tmin = 0.52. The final R1 and wR2(F2) were 0.0532 and 0.1322 (I > 2σ(I)), 0.0719 and 0.1438 (all data) for 1469 parameters and 40 restraints. CCDC-1420799.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 25410133.Notes and references
- (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (b) M. J. Hardie, Chem. Soc. Rev., 2010, 39, 516–527 RSC; (c) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760–1768 CrossRef CAS; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (f) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed.
- (a) M. Yoshizawa, M. Tamura and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 3874–3876 CrossRef CAS PubMed; (b) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed; (c) J. S. Mugridge, G. Szigethy, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 16256–16264 CrossRef CAS PubMed; (d) N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- (a) D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 6748–6751 CrossRef CAS PubMed; (b) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (c) M. D. Pluth, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2007, 46, 8587–8589 CrossRef CAS PubMed; (d) S. J. Lee, S.-H. Cho, K. L. Mulfort, D. M. Tiede, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 16828–16829 CrossRef CAS PubMed.
- (a) M. Ziegler, J. L. Brumaghim and K. N. Raymond, Angew. Chem., Int. Ed., 2000, 39, 4119–4121 CrossRef CAS; (b) M. Yoshizawa, T. Kusukawa, M. Fujita and K. Yamaguchi, J. Am. Chem. Soc., 2000, 122, 6311–6312 CrossRef CAS; (c) M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558–6559 CrossRef CAS PubMed; (d) V. M. Dong, D. Fiedler, B. Carl, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 14464–14465 CrossRef CAS PubMed.
- (a) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed; (b) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed; (d) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed.
- (a) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed; (b) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed; (c) S. P. Nolan, Acc. Chem. Res., 2010, 44, 91–100 CrossRef PubMed; (d) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- (a) V. K.-M. Au, K. M.-C. Wong, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 9076–9085 CrossRef CAS PubMed; (b) T. Zou, C. T. Lum, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed; (c) Y. Liu, T. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412–6414 RSC; (d) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- (a) S. Wei, X. Li, Z. Yang, J. Lan, G. Gao, Y. Xue and J. You, Chem. Sci., 2012, 3, 359–363 RSC; (b) B. N. Ahamed, R. Dutta and P. Ghosh, Inorg. Chem., 2013, 52, 4269–4276 CrossRef CAS PubMed; (c) A. Vellé, A. Cebollada, M. Iglesias and P. J. Sanz Miguel, Inorg. Chem., 2014, 53, 10654–10659 CrossRef PubMed; (d) C. Mejuto, G. Guisado-Barrios, D. Gusev and E. Peris, Chem. Commun., 2015, 51, 13914–13917 RSC.
- X. Hu, Y. Tang, P. Gantzel and K. Meyer, Organometallics, 2003, 22, 612–614 CrossRef CAS.
- (a) A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed; (b) A. Rit, T. Pape, A. Hepp and F. E. Hahn, Organometallics, 2011, 30, 334–347 CrossRef CAS; (c) A. Rit, T. Pape and F. E. Hahn, Organometallics, 2011, 30, 6393–6401 CrossRef CAS; (d) R. Maity, A. Rit, C. Schulte to Brinke, C. G. Daniliuc and F. E. Hahn, Chem. Commun., 2013, 49, 1011–1013 RSC; (e) C. Segarra, G. Guisado-Barrios, F. E. Hahn and E. Peris, Organometallics, 2014, 33, 5077–5080 CrossRef CAS; (f) N. Sinha, F. Roelfes, A. Hepp, C. Mejuto, E. Peris and F. E. Hahn, Organometallics, 2014, 33, 6898–6904 CrossRef CAS.
- (a) M. V. Baker, D. H. Brown, R. A. Haque, B. W. Skelton and A. H. White, Dalton Trans., 2004, 3756–3764 RSC; (b) C. E. Willans, K. M. Anderson, P. C. Junk, L. J. Barbour and J. W. Steed, Chem. Commun., 2007, 3634–3636 RSC; (c) F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Chem. – Eur. J., 2008, 14, 10900–10904 CrossRef CAS PubMed; (d) D. Wang, B. Zhang, C. He, P. Wu and C. Duan, Chem. Commun., 2010, 46, 4728–4730 RSC; (e) C. Radloff, H.-Y. Gong, C. Schulte to Brinke, T. Pape, V. M. Lynch, J. L. Sessler and F. E. Hahn, Chem. – Eur. J., 2010, 16, 13077–13081 CrossRef CAS PubMed.
- (a) M. Tominaga, H. Masu and I. Azumaya, J. Org. Chem., 2009, 74, 8754–8760 CrossRef CAS PubMed; (b) M. Tominaga, K. Ohara, K. Yamaguchi and I. Azumaya, J. Org. Chem., 2014, 79, 6738–6742 CrossRef CAS PubMed; (c) M. Tominaga, N. Kunitomi, K. Katagiri and T. Itoh, Org. Lett., 2015, 17, 786–789 CrossRef CAS PubMed.
- (a) S. V. Lindeman and J. K. Kochi, Cryst. Growth Des., 2004, 4, 563–571 CrossRef CAS; (b) W. M. Reichert, J. D. Holbrey, R. P. Swatloski, K. E. Gutowski, A. E. Visser, M. Nieuwenhuyzen, K. R. Seddon and R. D. Rogers, Cryst. Growth Des., 2007, 7, 1106–1114 CrossRef CAS; (c) C. A. Hollis, L. R. Hanton, J. C. Morris and C. J. Sumby, Cryst. Growth Des., 2009, 9, 2911–2916 CrossRef CAS.
- (a) Q. Teng and H. V. Huynh, Chem. Commun., 2015, 51, 1248–1251 RSC; (b) J. C. C. Chen and I. J. B. Lin, J. Chem. Soc., Dalton Trans., 2000, 839–840 RSC; (c) S. Saito, T. Kobayashi, T. Makino, H. Yamaguchi, H. Muto, I. Azumaya, K. Katagiri and R. Yamasaki, Tetrahedron, 2012, 68, 8931–8936 CrossRef CAS; (d) H.-X. Liu, X. He and L. Zhao, Chem. Commun., 2014, 50, 971–974 RSC.
- (a) A. Ikeda, H. Udzu, Z. Zhong, S. Shinkai, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2001, 123, 3872–3877 CrossRef CAS PubMed; (b) H.-J. Kim, D. Moon, M. S. Lah and J.-I. Hong, Angew. Chem., Int. Ed., 2002, 41, 3174–3177 CrossRef CAS; (c) S. Hiraoka, K. Harano, T. Tanaka, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2003, 42, 5182–5185 CrossRef CAS PubMed; (d) S. Hiraoka, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2004, 126, 1214–1218 CrossRef CAS PubMed; (e) X.-P. Li, J.-Y. Zhang, M. Pan, S.-R. Zheng, Y. Liu and C.-Y. Su, Inorg. Chem., 2007, 46, 4617–4625 CrossRef CAS PubMed; (f) Y. Tsunoda, K. Fukuta, T. Imamura, R. Sekiya, T. Furuyama, N. Kobayashi and T. Haino, Angew. Chem., Int. Ed., 2014, 53, 7243–7247 CrossRef CAS PubMed.
- D. N. Chin, D. M. Gordon and G. M. Whitesides, J. Am. Chem. Soc., 1994, 116, 12033–12044 CrossRef CAS.
- (a) G. M. Sheldrick, SHELX-97, Program for the Solution and Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of 1H and 13C NMR spectra for all precursors and complexes, mass spectra for complexes 3 and 6, and crystallographic information files (CIFs) of complexes 3 and 6. The crystal structure depository numbers for 3 and 6 are CCDC 1420798 and 1420799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01860a |
| This journal is © The Royal Society of Chemistry 2016 |