DOI:
10.1039/C5CE01792K
(Paper)
CrystEngComm, 2016,
18, 222-229
Syntheses and characterization of aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes†
Received
8th September 2015
, Accepted 20th October 2015
First published on 11th November 2015
Abstract
Seven aryl-substituted pyrogallol[4]arenes and six aryl-substituted resorcin[4]arenes were synthesized through the acid catalyzed reaction of either pyrogallol or resorcinol with a specific alkoxybenzaldehyde. Single crystal X-ray data was obtained for all thirteen compounds. In order to determine the effect of the different pendent –R groups, four properties were investigated: π–π distance, inward tilt, twist angle, and the angle between the planes containing the pendent –R groups. Positioning of the –R groups, the carbon atom chain length of the –R groups, the number of upper-rim hydroxyl groups (resorcin[4]arene vs. pyrogallol[4]arene), and the number of substituted phenyl groups all influenced these four properties. The trends that develop are investigated and discussed.
Introduction
Due to their flexibility and bowl-shaped cavity, calix[4]arenes have garnered attention over the last forty years. They have found extensive applications in a range of fields due to their ability to act as host molecules for a variety of guest molecules.1 As a result of the considerable number of applications of calix[4]arenes, related hosts were synthesized, such as pyrogallol[4]arenes and resorcin[4]arenes (see Fig. 1). Pyrogallol[4]arenes and resorcin[4]arenes imitate the conformation and shape of calix[4]arenes; however, the possibility of more hydrogen bonding due to the presence of more hydroxyl groups on the upper-rim (eight for resorcin[4]arenes and twelve for pyrogallol[4]arenes) has led to new chemistry and to supramolecular architectures such as metal-seamed dimers and hexamers.2,3
 |
| | Fig. 1 Schematic structures of (a) pyrogallol[4]arene and (b) resorcin[4]arene. | |
Niedel and Vogel led the way in the 1940s with research in resorcin[4]arenes. Their work concentrated on the reactions of resorcinol with aliphatic aldehydes. These reactions resulted in the formation of the all-cis cone stereoisomer (see Fig. 2).4,5
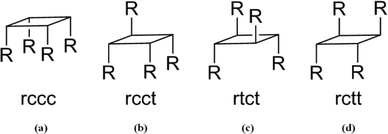 |
| | Fig. 2 Possible orientations of the –R groups (a) rccc, (b) rcct, (c) rtct, and (d) rctt. Reference group is the front, left group. | |
Later research by Hoegberg exposed three more possible conformers of resorcin[4]arenes: boat, saddle, and chair.6 The orientations of the pendent –R groups for resorcin[4]arene are given the nomenclature of rccc (cone), rcct (partial cone), rcct (saddle), and rctt (chair) and they describe the orientation of the aliphatic or aryl –R group (see Fig. 2). The reference point, r, is followed by stereochemical positions cis, c, or trans, t, going counterclockwise around the molecule. Vogel undertook further studies on the conformers of resorcin[4]arenes with variable temperature 1H NMR studies. It was determined that the kinetic product was the chair isomer while the cone isomer was the thermodynamic product.6
Pyrogallol[4]arenes and resorcin[4]arenes have different bonding interactions and structure.7 Therefore, manipulation of the pyrogallol[4]arene and resorcin[4]arene conformers is most likely different from that with calix[4]arenes. For instance, for pyrogallol[4]arenes it was determined that in aprotic solvent the chair conformation was preferred, but in protic solvent the boat conformation was favored.8 It has been hypothesized that the pendent –R group also might have an impact on the resulting conformation.9 A good deal of information is known about the synthesis pathways for pyrogallol[4]arenes and resorcin[4]arenes; however, not much is known about the properties and interactions that govern these reactions. Thus, characterization and the creation of a database of modified pyrogallol[4]arenes and resorcin[4]arenes is needed to provide insight into the interactions involved with these molecules and their adaptability and flexibility to specific applications. Accordingly, the work herein was carried out to uncover the properties and trends that arise from varying the phenyl substituent on pyrogallol[4]arenes and resorcin[4]arenes.
Several studies have already been carried out with aryl-substituted C-phenylpyrogallol[4]arenes10–13 and C-phenylresorcin[4]arenes.14 These have all produced the chair conformer unless the hydroxyl group has been alkylated (see Table 1). Additionally, studies with C-fluorophenyl-pyrogallol[4]arene, C-chlorophenylpyrogallol[4]arene, and C-bromophenylpyrogallol[4]arene have been completed (see Table 1).15 The twist angle (the degree of rotation between two eclipsed benzene ring substituents) is greatest and smallest with bromophenyl and fluorophenyl substituents respectively. Furthermore, it was found that temperature also played a part in influencing the twist angle. The twist angle decreased as the temperature decreased from reflux to room temperature.
Table 1 Previously synthesized aryl-substituted pyrogallol[4]arenes
| Pendent –R group |
Solvent system |
| Phenyl10a |
DMF |
| Biphenyl13 |
DMF |
| Naphthyl11 |
Methanol, pyrazine |
| 4-Cyanophenyl15 |
DMSO |
| 4-Fluorophenyl15 |
DMSO |
| 4-Chlorophenyl15 |
DMSO |
| 4-Bromophenyl15 |
DMSO |
| 4-Hydroxyphenyl10b |
DMSO |
Herein seven aryl-substituted pyrogallol[4]arenes and six aryl-substituted resorcin[4]arenes have been synthesized and single crystal X-ray data for all thirteen structures has been collected. Both the pendent –R groups and whether the molecule is a pyrogallol[4]arene or resorcin[4]arene affect several properties of the resulting structures, including the π–π distance between pendent –R groups, the inward tilt of the pendent –R groups, the twist angle of the pendent –R groups, and the angle between the planes containing the pendent –R groups (ABP). The trends are investigated and discussed in detail (Table 2).
Experimental
Reagents and solvents were obtained commercially and used without additional purification.
Table 2 Structures with corresponding name and –R group. –R groups attaches to bridging –CH linker through the top (top left, if more than one phenyl group) carbon atom
| Structure |
Name |
–R group |
| 1 |
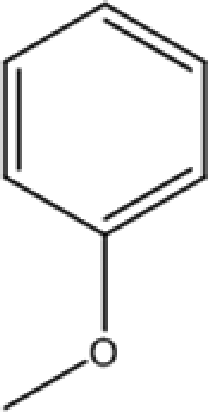
C-4-Methoxyphenylpyrogallol[4]arene |

|
| 2 |
C-2-Methoxyphenylpyrogallol[4]arene |
|
| 3 |
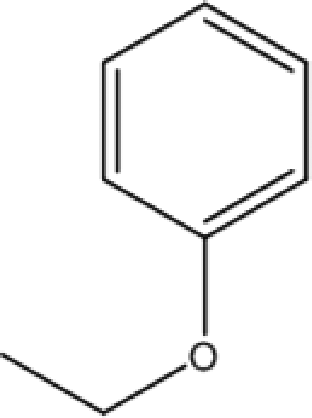
C-4-Ethoxyphenylpyrogallol[4]arene |
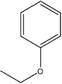
|
| 4 |
C-4-Propoxyphenylpyrogallol[4]arene |
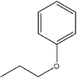
|
| 5 |
C-4-Butoxyphenylpyrogallol[4]arene |

|
| 6 |
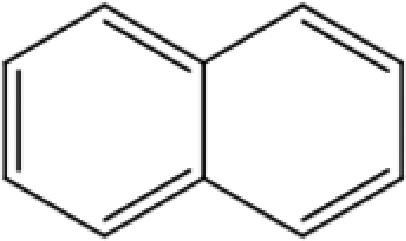
C-1-Naphthylpyrogallol[4]arene |

|
| 7 |
C-4-Methoxy-1-naphthylpyrogallol[4]arene |
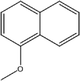
|
| 8 |
C-4-Methoxyphenylresorcin[4]arene |

|
| 9 |
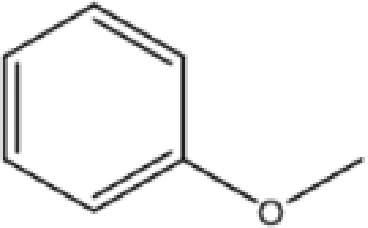
C-3-Methoxyphenylresorcin[4]arene |

|
| 10 |
C-2-Methoxyphenylresorcin[4]arene |

|
| 11 |
C-4-Ethoxyphenylresorcin[4]arene |

|
| 12 |
C-4-Isopropoxyphenylresorcin[4]arene |
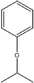
|
| 13 |
C-1-Naphthylresorcin[4]arene |

|
Synthesis of C-4-methoxyphenylpyrogallol[4]arene (1)
4-Methoxybenzaldehyde.
Into 5 grams of 4-hydroxybenzaldehyde, 125 mL of DMF was added. The reaction was stirred until all 4-hydroxybenzaldehyde dissolved. To the reaction, 1.97 grams of sodium hydride was added and the solution was stirred at room temperature for ten minutes. To the solution, 3.05 mL of iodoethane was added and the solution was stirred at room temperature for one hour. The reaction was quenched with methanol then all solvent was rotovapped off. The product was washed with a 50/50 water/chloroform mixture. Since product dissolves in chloroform, the chloroform layer was removed and rotovapped off. The remaining liquid was dried with magnesium sulfate, filtered, and all remaining solvent was evaporated off yielding 8.3 grams of orange liquid.
C-4-Methoxyphenylpyrogallol[4]arene.
To a round bottom flask, 3.89 grams of pyrogallol and 50 mL of ethanol were added. The solution was stirred until the pyrogallol dissolved. To the solution, 3.75 mL of 4-methoxybenzaldehyde was added followed by 0.5 mL of concentrated hydrochloric acid. The solution was heated to 90 °C and refluxed for 8 hours. The solution was filtered and the powder dried yielding 1.8 grams of white precipitate. Colourless, plate-shaped crystals were obtained by dissolving the powder in DMSO and allowing the solution to slowly evaporate.
Synthesis of structures 2–13
Synthesis of structures 2–13 were synthesized with a similar method as structure 1. Complete synthesis information is described in ESI.†
Crystallography
Single crystal X-ray data for structures 2, 8, 10, and 13 were collected at 173 K on a Bruker Apex II CCD diffractometer using a CuKα radiation source (1.54178 Å). Data for all other cocrystals were collected at 100 K or 173 K on a Bruker Apex II CCD diffractometer, using a MoKα radiation source (0.71073 Å).
Cocrystal 1.
C76H104O28S8, M = 1774.15, colourless prism, a = 16.0033(7) Å, b = 11.5923(5) Å, c = 22.053(1) Å, β = 94.549(2)°, space group P21/c, V = 4078.3(3) Å3, Z = 2, Dc = 1.363 g cm−3, F000 = 1776, MoKα radiation, λ = 0.71073 Å, T = 173 K, 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 413 reflections collected. Final GooF = 1.08, R1 = 0.070, wR2 = 0.108, R indices based on reflections with I > 2σ(I) (refinement on F2), 553 parameters, 54 restraints. Lp and absorption corrections applied, μ = 0.297 mm−1.
413 reflections collected. Final GooF = 1.08, R1 = 0.070, wR2 = 0.108, R indices based on reflections with I > 2σ(I) (refinement on F2), 553 parameters, 54 restraints. Lp and absorption corrections applied, μ = 0.297 mm−1.
Cocrystal 2.
C76H123.8O26S10, M = 1774.15, colourless prism, a = 13.3120(3) Å, b = 17.0039(4) Å, c = 22.7649(5) Å, α = 105.340(1)°, β = 102.612(1)°, γ = 106.369(1)°, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , V = 45.22.76(18) Å3, Z = 2, Dc = 1.303 g cm−3, F000 = 1896, CuKα radiation, λ = 1.54178 Å, T = 173 K, 14967 reflections collected. Final GooF = 2.47, R1 = 0.232, wR2 = 0.564, R indices based on reflections with I > 2σ(I) (refinement on F2), 1021 parameters, 102 restraints. Lp and absorption corrections applied, μ = 2.85 mm−1.
, V = 45.22.76(18) Å3, Z = 2, Dc = 1.303 g cm−3, F000 = 1896, CuKα radiation, λ = 1.54178 Å, T = 173 K, 14967 reflections collected. Final GooF = 2.47, R1 = 0.232, wR2 = 0.564, R indices based on reflections with I > 2σ(I) (refinement on F2), 1021 parameters, 102 restraints. Lp and absorption corrections applied, μ = 2.85 mm−1.
Cocrystal 3.
C72H92O22S6, M = 1501.82, colourless plate, a = 10.3128(4) Å, b = 13.6528(5) Å, c = 15.1819(6) Å, α = 113.311(2)°, β = 105.470(2)°, γ = 95.699(2)°, space group P, V = 1840.6(1) Å3, Z = 1, Dc = 1.355 g cm−3, F000 = 796, MoKα radiation, λ = 0.71073 Å, T = 173 K, 8668 reflections collected. Final GooF = 1.02, R1 = 0.073, wR2 = 0.102, R indices based on reflections with I > 2σ(I) (refinement on F2), 465 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.26 mm−1.
Cocrystal 4.
C176H272O56S24, M = 4053.38, pink prism, a = 13.980(1) Å, b = 16.337(2) Å, c = 24.278(2) Å, α = 77.851(1)°, β = 73.707(1)°, γ = 73.719(1)°, space group P, V = 5056.3(9) Å3, Z = 1, Dc = 1.331 g cm−3, F000 = 2160, MoKα radiation, λ = 0.71073 Å, T = 173 K, 20582 reflections collected. Final GooF = 1.07, R1 = 0.171, wR2 = 0.265, R indices based on reflections with I > 2σ(I) (refinement on F2), 1190 parameters, 119 restraints. Lp and absorption corrections applied, μ = 0.33 mm−1.
Cocrystal 5.
C92H144O28S12, M = 2082.79, colourless prism, a = 13.564(1) Å, b = 14.191(1) Å, c = 15.937(1) Å, α = 110.429(4)°, β = 109.634(4)°, γ = 92.884(3)°, space group P, V = 2658(3) Å3, Z = 1, Dc = 1.301 g cm−3, F000 = 1112, MoKα radiation, λ = 0.71073 Å, T = 173 K, 10615 reflections collected. Final GooF = 1.02, R1 = 0.121, wR2 = 0.193, R indices based on reflections with I > 2σ(I) (refinement on F2), 662 parameters, 48 restraints. Lp and absorption corrections applied, μ = 0.32 mm−1.
Cocrystal 6.
C96H132O26S14, M = 2150.86, colourless prism, a = 12.973(2) Å, b = 14.665(2) Å, c = 15.949(2) Å, α = 97.687(2)°, β = 112.271(2)°, γ = 98.894(2)°, space group P, V = 2712.0(7) Å3, Z = 1, Dc = 1.317 g cm−3, F000 = 1140, MoKα radiation, λ = 0.71073 Å, T = 173 K, 12058 reflections collected. Final GooF = 1.06, R1 = 0.139, wR2 = 0.334, R indices based on reflections with I > 2σ(I) (refinement on F2), 615 parameters, 36 restraints. Lp and absorption corrections applied, μ = 0.35 mm−1.
Cocrystal 7.
C86H99.8O23S7, M = 1725.88, colourless prism, a = 12.3525(6) Å, b = 13.8124(8) Å, c = 15.0196(8) Å, α = 112.326(2)°, β = 112.595(2)°, γ = 93.556(2)°, space group P, V = 2122.7(2) Å3, Z = 1, Dc = 1.350 g cm−3, F000 = 912, MoKα radiation, λ = 0.71073 Å, T = 173 K, 7073 reflections collected. Final GooF = 1.83, R1 = 0.165, wR2 = 0.434, R indices based on reflections with I > 2σ(I) (refinement on F2), 579 parameters, 66 restraints. Lp and absorption corrections applied, μ = 0.26 mm−1.
Cocrystal 8.
C152H216O44S20, M = 1537.97, colourless prism, a = 10.6715(3) Å, b = 16.8763(5) Å, c = 24.6079(7) Å, α = 97.900(1)°, β = 102.264(1)°, γ = 99.408(1)°, space group P, V = 4203.7(2) Å3, Z = 1, Dc = 1.339 g cm−3, F000 = 1800, CuKα radiation, λ = 1.54178 Å, T = 173 K, 14941 reflections collected. Final GooF = 1.09, R1 = 0.138, wR2 = 0.249, R indices based on reflections with I > 2σ(I) (refinement on F2), 1031 parameters, 30 restraints. Lp and absorption corrections applied, μ = 3.01 mm−1.
Cocrystal 9.
C72H96O20S8, M = 1537.98, colourless plate, a = 12.529(1) Å, b = 13.245(1) Å, c = 13.638(1) Å, α = 112.439(1)°, β = 109.559(1)°, γ = 92.935(1)°, space group P, V = 1929.0(3) Å3, Z = 1, Dc = 1.324 g cm−3, F000 = 816, MoKα radiation, λ = 0.71073 Å, T = 100 K, 8543 reflections collected. Final GooF = 1.03, R1 = 0.060, wR2 = 0.104, R indices based on reflections with I > 2σ(I) (refinement on F2), 482 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.30 mm−1.
Cocrystal 10.
C80H120O24S12, M = 1850.48, colourless prism, a = 13.6701(2) Å, b = 16.5031(3) Å, c = 21.1363(3) Å, β = 99.896(1)°, space group P21/n, V = 4697.4(1) Å3, Z = 2, Dc = 1.308 g cm−3, F000 = 1968, CuKα radiation, λ = 1.54178 Å, T = 173 K, 8608 reflections collected. Final GooF = 3.29, R1 = 0.206, wR2 = 0.602, R indices based on reflections with I > 2σ(I) (refinement on F2), 552 parameters, 66 restraints. Lp and absorption corrections applied, μ = 3.16 mm−1.
Cocrystal 11.
C156H220O42S18, M = 953.01, colourless plate, a = 13.887(2) Å, b = 15.638(2) Å, c = 23.205(3) Å, α = 99.446(2)°, β = 101.639(2)°, γ = 114.753(2)°, space group P, V = 4304(1) Å3, Z = 1, Dc = 1.290 g cm−3, F000 = 1780, MoKα radiation, λ = 0.71073 Å, T = 100 K, 14853 reflections collected. Final GooF = 1.02, R1 = 0.131, wR2 = 0.196, R indices based on reflections with I > 2σ(I) (refinement on F2), 1019 parameters, 32 restraints. Lp and absorption corrections applied, μ = 0.30 mm−1.
Cocrystal 12.
C160H224O40S16, M = 3300.35, colourless prism, a = 12.108(1) Å, b = 13.592(1) Å, c = 26.446(3) Å, α = 88.602(1)°, β = 85.738(1)°, γ = 83.463(1)°, space group P, V = 4311.5(7) Å3, Z = 1, Dc = 1.270 g cm−3, F000 = 1760, MoKα radiation, λ = 0.71073 Å, T = 100 K, 17182 reflections collected. Final GooF = 1.02, R1 = 0.100, wR2 = 0.120, R indices based on reflections with I > 2σ(I) (refinement on F2), 1005 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.27 mm−1.
Cocrystal 13.
C128H108O8N12, M = 1942.26, colourless plate, a = 13.0650(3) Å, b = 14.1576(3) Å, c = 15.2003(3) Å, α = 72.404(1)°, β = 76.963(1)°, γ = 75.955(1)°, space group P, V = 2564.45(9) Å3, Z = 1, Dc = 1.258 g cm−3, F000 = 1024, CuKα radiation, λ = 1.54178 Å, T = 173 K, 8990 reflections collected. Final GooF = 1.054, R1 = 0.062, wR2 = 0.175, R indices based on reflections with I > 2σ(I) (refinement on F2), 665 parameters, 0 restraints. Lp and absorption corrections applied, μ = 0.63 mm−1.
Results
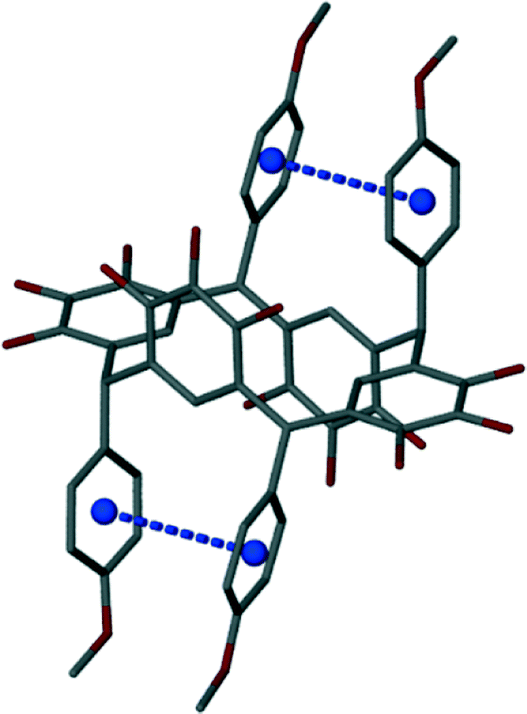
All synthesized structures form the chair conformer pack in a bilayer arrangement. Four measurements are used to describe and compare the structures of the aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes: π–π distance, the tilt inward, the twist angle, and the angle between the plane of the pendent –R groups (see Tables 3 and 4). (Note: there are two sets of pendent –R groups, four pendent –R groups total. Some of the molecules are symmetric through a C2h plane so the two groups are equal and thus only one set of measurements is given.) First, the π–π distance is the distance measured from the calculated centroids of the phenyl groups of the pendent –R groups (see Fig. 3). This measurement describes the degree the pendent –R groups are rotated away from each other and is used with the tilt distance to express how much the pendent –R groups are tilted inwards (see Fig. 4). Tilt distance is the difference between the two C–C distances (see Fig. 5). The first C–C distance is the distance between the two C4 carbon atoms of the phenyl groups of the –R groups and the second C–C distance is the distance between the two C1 carbon atoms of the phenyl groups of the –R groups (see Fig. 5). The twist angle is found using equation 1. Angle 1 is the angle that is made up by the points C1, C2, and C3 (see Fig. 6). Angle 2 is the angle that is made up by the points C3, C4, and C5 (see Fig. 6). Along with the angle between the planes of the eclipsed pendent –R groups, the twist angle describes the sterics and torsion between the two groups. The angle between the planes (ABP) of the eclipsed pendent –R groups is calculated with the MPLA command in X-Seed.16 The command creates two planes for the two phenyl groups and then calculates the angle between.| | | Twist angle = (90° − angle 1) + (90° − angle 2) | (1) |
Table 3 π–π distance and inward tilt of aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes
| Structure |
π–π distance (Å) |
C–C distance 1 (Å) |
C–C distance 2 (Å) |
Tilt inwarda (Å) |
|
Tilt inward is calculated as the difference between C–C distance 2 and C–C distance 1.
|
| 1 |
4.59 |
4.29 |
4.88 |
0.59 |
| 2 |
4.63 |
4.47 |
4.87 |
0.40 |
|
|
4.42 |
4.01 |
4.81 |
0.80 |
| 3 |
4.68 |
4.45 |
4.91 |
0.46 |
| 4 |
4.90 |
4.85 |
4.96 |
0.11 |
|
|
4.90 |
4.84 |
4.96 |
0.12 |
| 5 |
4.83 |
4.77 |
4.96 |
0.19 |
| 6 |
4.29 |
3.89 |
4.74 |
0.85 |
| 7 |
4.40 |
4.14 |
4.74 |
0.60 |
| 8 |
4.34 |
3.93 |
4.77 |
0.84 |
|
|
4.34 |
3.93 |
4.77 |
0.84 |
| 9 |
4.74 |
4.49 |
4.98 |
0.49 |
| 10 |
4.43 |
4.08 |
4.84 |
0.76 |
| 11 |
4.54 |
4.30 |
4.84 |
0.54 |
|
|
4.46 |
4.19 |
4.81 |
0.62 |
| 12 |
4.59 |
4.46 |
4.82 |
0.36 |
|
|
5.10 |
5.12 |
5.06 |
−0.06 |
| 13 |
4.68 |
4.56 |
4.87 |
0.31 |
Table 4 Twist angles and angle between the planes of eclipsed –R groups for aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes
| Structure |
Angle 1 (°) |
Angle 2 (°) |
90° − angle 1 (°) |
90° − angle 2 (°) |
Twist angle (°) |
ABP (°) (esd) |
| 1 |
85.4 |
85.5 |
4.6 |
4.5 |
9.1 |
21.57 (0.14) |
| 2 |
87.1 |
86.3 |
2.9 |
3.7 |
6.6 |
10.26 (1.14) |
|
|
86.6 |
85.3 |
3.4 |
4.7 |
8.1 |
16.53 (1.15) |
| 3 |
85.6 |
88.0 |
4.4 |
2.0 |
6.4 |
19.12 (0.13) |
| 4 |
91.6 |
122 |
−1.6 |
−32 |
−33.8 |
67.09 (0.55) |
|
|
91.6 |
90.2 |
−1.6 |
−0.2 |
−1.8 |
67.11 (0.55) |
| 5 |
84.8 |
89.1 |
5.2 |
0.9 |
6.1 |
30.52 (0.40) |
| 6 |
83.0 |
83.6 |
7.0 |
6.4 |
13.4 |
21.31 (0.18) |
| 7 |
80.1 |
88.8 |
9.9 |
1.2 |
11.1 |
16.27 (0.34) |
| 8 |
83.3 |
85.2 |
6.7 |
4.8 |
11.5 |
22.75 (0.26) |
|
|
83.0 |
85.2 |
7.0 |
4.8 |
11.8 |
22.63 (0.26) |
| 9 |
85.7 |
86.9 |
4.3 |
3.1 |
7.4 |
49.79 (0.08) |
| 10 |
84.3 |
84.5 |
5.7 |
5.5 |
11.2 |
16.42 (0.35) |
| 11 |
83.8 |
88.0 |
6.2 |
2.0 |
8.2 |
22.60 (0.29) |
|
|
81.4 |
88.0 |
1.2 |
8.6 |
9.8 |
21.19 (0.25) |
| 12 |
83.8 |
89.3 |
6.2 |
0.7 |
6.9 |
21.63 (0.14) |
|
|
90.1 |
90.4 |
−0.1 |
−0.4 |
−0.5 |
45.15 (0.10) |
| 13 |
86.4 |
87.0 |
3.6 |
3.0 |
6.6 |
14.16 (0.08) |
 |
| | Fig. 3 The π–π distance (dashed blue bond) and the calculated centroids (blue atoms). Hydrogen atoms are removed for clarity. | |
 |
| | Fig. 4 Difference between pendent –R groups tilting inwards and outwards. Hydrogen atoms are removed for clarity. | |
 |
| | Fig. 5 C4 C–C distance (dashed blue bond) and C1 C–C distance (dashed green bond). C4 atoms are blue while C1 atoms are green. Hydrogen atoms are removed for clarity. | |
 |
| | Fig. 6 Points C1 and C2 that make up angle 1 (blue atoms) and points C4 and C5 that make up angle 2 (green atoms). C3 is used to find both angles and is the orange atom. Hydrogen atoms are removed for clarity. | |
Discussion
Several trends arose from the change in pendent –R group and the change from pyrogallol[4]arene to resorcin[4]arene. A summary of the trends is found at the end of this section in Table 7 (for compound 13, keep in mind that it is crystallized in pyridine, not DMSO like all other compounds). First examined is π–π distance. With the pyrogallol[4]arene-based compounds (excluding naphthalene-based) (2 and 3), the π–π distance is greater than in resorcin[4]arene-based compounds (8, 10, and 11) (see Table 5). There are two different trends for π–π distance in pyrogallol[4]arene and resorcin[4]arene compounds that have naphthyl as the –R group. In the pyrogallol[4]arene compounds, the pyrogallol[4]arene with a para-methoxyphenyl –R group has a greater π–π distance than the pyrogallol[4]arene with a naphthyl –R group (5) (4.59 Å and 4.29 Å, respectively). Furthermore, the C-1-naphthylpyrogallol[4]arene has a smaller π–π distance than C-4-methoxy-1-naphthylpyrogallol[4]arene (6) (4.29 Å and 4.40 Å, respectively). In the resorcin[4]arene compound, the trend is reversed; the resorcin[4]arene with a para-methoxyphenyl –R group (7) has a smaller π–π distance than the resorcin[4]arene with a naphthyl –R group (13) (4.34 Å and 4.68 Å, respectively). Both the pyrogallol[4]arene and resorcin[4]arene compounds have the same trend in π–π distance when the alkoxy group is extended from one carbon atom (methoxyphenyl) to three carbon atoms (propoxyphenyl or isopropoxyphenyl). As the alkoxy group increases in carbon atoms, the π–π distance also increases (methoxy to ethoxy to propoxy: 4.59 Å to 4.68 Å to 4.90 Å for pyrogallol[4]arene compounds and 4.34 Å to 4.46 Å, 4.54 Å to 4.59 Å, 5.10 Å for resorcin[4]arene compounds). Finally, both pyrogallol[4]arene and resorcin[4]arene compounds have the same trend in π–π distance when the position of the methoxy substituent is changed. With the meta-methoxyphenyl substituent, the π–π distance is the greatest, followed by the ortho-methoxyphenyl substituent, and the para-methyoxyphenyl substituent has the smallest π–π distance (para, ortho, meta: 4.59 Å, and 4.64 Å (4.38 Å, second value) for pyrogallol[4]arene compounds and 4.34 Å, 4.43 Å, and 4.74 Å for resorcin[4]arene compounds).
Table 5 Comparison of π–π distances and inward tilt distances for pyrogallol[4]arenes vs. resorcin[4]arenes
| Structure |
–R group |
π–π distance (Å) |
Tilt inward (Å) |
| 1 |
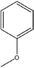
|
4.59 |
0.59 |
| 2 |

|
4.63 |
0.40 |
|
|
|
4.42 |
0.80 |
| 3 |

|
4.68 |
0.46 |
| 8 |

|
4.34 |
0.84 |
|
|
|
4.34 |
0.84 |
| 10 |
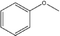
|
4.43 |
0.76 |
| 11 |

|
4.54 |
0.54 |
|
|
|
4.46 |
0.62 |
The second measurement inspected is the tilt inward. The smaller the number, the more the pendent –R groups are tilted inward. Pyrogallol[4]arene compounds (2 and 3) have pendent –R groups that tilt inward more than the pendent –R groups of resorcin[4]arene compounds (8, 10, and 11) (see Table 5). Once again, with naphthyl as the –R group, the pyrogallol[4]arene compounds (5) and the resorcin[4]arene compounds (8 and 13) have opposite trends. The tilt inward for pendent –R groups on pyrogallol[4]arene compounds is greater in C-4-methyoxyphenylpyrogallol[4]arene and smaller in C-1-naphthylpyrogallol[4]arene (0.59 Å and 0.85 Å, respectively). The opposite trend is found for resorcin[4]arene compounds; the tilt inward for pendent –R groups is greater in C-1-naphthylresorcin[4]arene and smaller in C-4-methyoxyphenylresorcin[4]arene (0.31 Å and 0.84 Å, respectively). With naphthyl and 4-methoxy-1-naphthyl substituents, the naphthyl substituted compound (5) has a smaller tilt inward than the 4-methoxy-1-naphthyl substituted compound (6) (0.60 Å and 0.85 Å, respectively). In terms of the number of carbon atoms in the pendent –R group, pyrogallol[4]arene and resorcin[4]arene compounds have the same trend. When the alkoxy group is extended from one carbon atom (methoxy) to three carbon atoms (propoxy or isopropoxy), the tilt of the pendent –R group inward is smallest for the methoxyphenyl substituted compounds (methoxy to ethoxy to propoxy: 0.59 Å to 0.46 Å to 0.11 Å for pyrogallol[4]arene compounds and 0.84 Å to 0.54 Å, 0.62 Å to 0.36 Å, −0.06 Å for resorcin[4]arene compounds). Finally, in regard to ortho, meta, and para positions of the substituents, ortho substituted compounds have –R groups that tilt more inwards than para substituted compounds. Meta substituted compounds have the greatest tilt inwards of the pendent –R groups (para, ortho, meta: 0.59 Å, 0.52 Å (0.75 Å, second value) for pyrogallol[4]arene compounds and 0.84 Å, 0.76 Å, 0.49 Å for resorcin[4]arene compounds).
Next examined is the twist angle. The greater the twist angle, the more inward the pendent –R groups are tilted. The twist angle is greater in resorcin[4]arene compounds than in pyrogallol[4]arene compounds (see Table 6). Also, naphthyl substituted pyrogallol[4]arenes (5) have greater twist angles than 4-methoxylphenyl substituted pryrogallol[4]arenes (13.4° and 9.1°, respectively). The reverse is true for resorcin[4]arene compounds (8 and 13). Naphthyl substituted resorcin[4]arenes have smaller twist angles than 4-methoxylphenyl substituted resorcin[4]arenes (6.6° and 11.5°, 11.6°, respectively). In regards to naphthyl and 4-methyoxy-1-naphthyl substituents, the naphthyl-substituted pyrogallol[4]arene (5) has a greater twist angle than the 4-methyoxy-1-naphthyl-substituted pyrogallol[4]arene (7) (13.4° and 11.1°, respectively). For both the pyrogallol[4]arene and resorcin[4]arene compounds, when the substituted pendent –R groups expand from methoxylphenyl to propoxyphenyl or isopropoxy phenyl, the twist angle decreases (methoxy to ethoxy to propoxy: 9.1° to 6.4° to −33.8°, −1.8° for pyrogallol[4]arene compounds and 11.5°, 11.8° to 8.2°, 9.8° to −0.5°, 6.9° for resorcin[4]arene compounds). Ortho-substituted pyrogallol[4]arenes have the greatest twist angle, followed by para-substituted compounds (para, ortho: 9.1°, 10.4°, (8.4°, second value). For resorcin[4]arene compounds, the meta-substituted compounds have the smallest twist angle and the ortho- and para-substituted compounds have similar twist angles (meta, ortho, para: 7.4°, 11.2°, 11.5°(11.6°)).
Table 6 Comparison of twist angle and ABP for pyrogallol[4]arenes vs. resorcin[4]arenes
| Structure |
–R group |
Twist angle (°) |
ABP (°) |
| 1 |

|
9.1 |
21.57 |
| 2 |

|
6.6 |
10.26 |
|
|
|
8.1 |
16.53 |
| 3 |

|
6.4 |
19.12 |
| 8 |
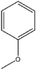
|
11.5 |
22.75 |
|
|
|
11.8 |
22.63 |
| 10 |

|
11.2 |
16.42 |
| 11 |

|
8.2 |
22.60 |
|
|
|
9.8 |
21.19 |
Finally, the last measurement looked at is the angle between the planes of the eclipsed pendent –R groups (ABP). Resorcin[4]arene compounds (8, 10, and 11) have greater ABPs than pyrogallol[4]arene compounds (2) (see Table 6). Pyrogallol[4]arenes and resorcin[4]arenes have the same trend dealing with the ABP of naphthyl –R groups. In both the pyrogallol[4]arenes and resorcin[4]arenes, the naphthyl-substituted compound (6 and 13 respectively) has a smaller ABP than the methyoxyphenyl-substituted compound (8) (pyrogallol[4]arene: 21.31° and 21.57° respectively, resorcin[4]arene: 14.16° and 22.63°, 22.75°, respectively). The naphthyl-substituted compounds (6) have a greater twist angle than the 4-methoxy-1-naphthyl-substituted compounds (7) (21.31° and 16.27°, respectively). When the substituted groups are expanded from one carbon atom (methyoxyphenyl) to three carbon atoms (propoxy or isopropoxy), the ABP decreases from methoxyphenyl to ethoxyphenyl but increases from ethoxyphenyl to propoxyl/isopropoxyphenyl (methoxy to ethoxy to propoxy: 21.57° to 19.12° to 67.09°, 67.11° for pyrogallol[4]arene compounds and 22.63°, 22.75° to 21.19°, 22.60° to 21.63°, 45.15° for resorcin[4]arene compounds). Ortho-substituted compounds have the smallest ABP, followed by para-substituted compounds, and meta-substituted compounds have the largest ABP ((ortho, para, meta: 12.45° (14.47°, second value), 21.57° for pyrogallol[4]arene compounds and 16.42°, 22.63°(22.75°), 49.79° for resorcin[4]arene compounds). A summary of all the trends discovered is found in Table 7.
Table 7 Trends for π–π distance, inward tilt, twist angle, and the angle between the planes (ABP) containing the pendent –R groups for all aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes
|
|
Measurement |
| Properties |
π–π distance |
Inward tilt |
Twist angle |
ABP |
| Pyrogallol[4]arene (pyro) vs. resorcin[4]arene (res) |
Pyro > res |
Pyro < res |
Pyro < res |
Pyro < res |
| Naphthyl (naph) vs. methoxyphenyl (met) |
Pyro met > naph res met < naph |
Pyro met < naph res met > naph |
Pyro met < naph res met > naph |
Pyro met > naph res met > naph |
| Methoxyl (1), ethoxyl (2), vs. propoxyphenyl (3) |
1 < 2 < 3 |
1 > 2 > 3 |
1 > 2 > 3 |
1 > 2 < 3 |
|
Ortho (o) vs. meta (m) vs. para (p) |
p < o < m |
p > o > m |
m < o ≈ p |
o < p < m |
In order to compare these current results to previously published crystal structures of aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes, similar examinations were performed with the previously reported structures. The results can be found in Table 8. Comparing the substituted pyrogallol[4]arenes to the substituted resorcin[4]arenes, with a phenyl or fluoro –R group, the pyrogallol[4]arenes have a greater π–π distances and angle between the planes than the resorcin[4]arenes. However, the substituted resorcin[4]arenes have a greater inward tilt and twist angle than the substituted pyrogallol[4]arenes. This is similar to the trends found for alkoxy substituted pyrogallol[4]arenes and resorcin[4]arenes except for the angle between the planes. For chloro –R groups, the pyrogallol[4]arenes have a greater inward tilt and twist angle than the resorcin[4]arenes and the resorcin[4]arenes have a greater π–π distances and angle between the planes. These trends are dissimilar to the trends found in the alkoxy substituted pyrogallol[4]arenes except for the angle between the planes (see Table 9 for a summary of the trends).
Table 8 π–π distance, inward tilt, twist angle, and angle between the plane of previously reported aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes
| Structure |
π–π distance (Å) |
Tilt inward (Å) |
Twist angle (°) |
ABP (°) |
|
Two reported structures.
Asymmetric structure, two set of phenyl rings.
|
|
C-Phenylpyrogallol[4]arene10a |
4.84 |
0.24 |
0.8 |
41.09 |
|
C-4-Cyanophenylpyrogallol[4]arene15a |
4.53 |
0.62 |
8.8 |
33.60 |
|
C-4-Cyanophenylpyrogallol[4]arene15a |
4.33 |
0.81 |
15.5 |
21.18 |
|
C-4-Chlorophenylpyrogallol[4]arene15 |
4.33 |
0.80 |
13.3 |
18.78 |
|
C-4-Bromophenylpyrogallol[4]arene15a |
4.50 |
0.64 |
7.9 |
20.05 |
|
C-4-Bromophenylpyrogallol[4]arene15a |
4.57 |
0.59 |
8.3 |
15.28 |
|
C-4-Fluorophenylpyrogallol[4]arene15 |
4.25 |
0.95 |
14.9 |
25.34 |
|
C-Phenylresorcin[4]arene14a |
4.17 |
1.12 |
16.8 |
23.30 |
|
C-4-Chlorophenylresorcin[4]arene14cb |
4.47, 4.68 |
0.35, 0.68 |
4.6, 8.5 |
23.37, 33.57 |
|
C-4-Fluorophenylresorcin[4]arene14c |
4.18 |
1.03 |
16 |
24.11 |
Table 9 Trends for π–π distance, inward tilt, twist angle, and the angle between the planes (ABP) containing the pendent –R groups for previously reported aryl substituted pyrogallol[4]arenes and resorcin[4]arenes
|
|
Measurement |
| Properties |
π–π distance |
Inward tilt |
Twist angle |
ABP |
| Pyrogallol[4]arene (pyro) vs. resorcin[4]arene (res) |
Phenyl, fluoro pyro > res chloro pyro < res |
Phenyl, fluoro pyro < res chloro pyro > res |
Phenyl, fluoro pyro < res chloro pyro > res |
Phenyl, fluoro pyro > res chloro pyro < res |
| Chloro (Cl) vs. bromo (Br) vs. fluoro (F) |
F < Cl < Br |
Br < Cl < F |
F < Cl < Br |
F < Cl < Br |
When comparing the chloro-, fluoro-, and bromo-substituted pyrogallol[4]arenes and resorcin[4]arenes, several trends emerge. For π–π distances, twist angle, and angle between the planes the bromo-substituted pyrogallol[4]arenes and resorcin[4]arenes have the greatest values, followed by the chloro-substituted pyrogallol[4]arenes and resorcin[4]arenes, and the fluoro-substituted pyrogallol[4]arenes and resorcin[4]arenes have the smallest values. The opposite is true for the inward tilt. The bromo-pyrogallol[4]arenes and resorcin[4]arenes have the smallest inward tilt and the fluoro-substituted pyrogallol[4]arenes and resorcin[4]arenes have the greatest inward tilt (see Table 9 for a summary of the trends).
Conclusions
Thirteen new aryl-substituted pyrogallol[4]arenes and resorcin[4]arenes were synthesized. It was demonstrated that small changes in the substituted pendent –R group (positioning of alkoxy group and length of alkoxy group) led to structural changes and several trends arose in π–π distance, inward tilt, twist angle, and ABP.
Further studies are being undertaken to determine the effect of substitution on phenylpyrogallol[4]arenes. Longer alkoxy groups in different positions are being synthesized as only methoxyphenyl was done in all three (ortho, meta, and para) positions. Additionally, phenyl rings are being expanded to determine if anthracene and pyrene groups could be substituted. Furthermore, studies have been started to convert the chair conformer of all structures to the boat conformer. This is being attempted through refluxing, microwave synthesis, or changes in solvent system. With the boat conformers synthesized, these compounds will be used to create a library of metal-seamed dimeric and hexameric nano-capsules.
Acknowledgements
JLA thanks the NSF for funding this work.
Notes and references
- V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
-
(a) S. J. Dalgarno, G. W. V. Cave and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 45, 570–574 CrossRef CAS PubMed;
(b) O. V. Kulikov, M. M. Daschbach, C. R. Yamnitz, N. Rath and G. W. Gokel, Chem. Commun., 2009, 7497–7499 RSC.
-
(a) S. J. Dalgarno, N. P. Power, J. E. Warren and J. L. Atwood, Chem. Commun., 2008, 1539–1541 RSC;
(b) S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS;
(c) P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760–1768 CrossRef CAS;
(d) P. Jin, S. J. Dalgarno, J. E. Warren, S. J. Teat and J. L. Atwood, Chem. Commun., 2009, 3348–3350 RSC;
(e) R. M. McKinlay, G. W. V. Cave and J. L. Atwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5944–5948 CrossRef CAS PubMed.
- J. B. Niedel and H. J. Vogel, J. Am. Chem. Soc., 1940, 62, 2512–2514 CrossRef.
- B. O. Nilsson, Acta Chem. Scand., 1968, 22, 732–747 CrossRef CAS.
- A. G. S. Hoegberg, J. Am. Chem. Soc., 1980, 102, 6046–6050 CrossRef CAS.
- R. K. Castellano, D. M. Rudkevich and J. Rebek, J. Am. Chem. Soc., 1996, 118, 10002–10003 CrossRef CAS.
- C. Schiel, G. A. Hembury, V. V. Borovkov, M. Klaes, C. Agena, T. Wada, S. Grimme, Y. Inoue and J. Mattay, J. Org. Chem., 2005, 71, 976–982 CrossRef PubMed.
- L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Org. Chem., 1989, 54, 1305–1312 CrossRef CAS.
-
(a) J. P. Kass, C. H. Zambrano, M. Zeller, A. D. Hunter and E. E. Dueno, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2006, 62, 3179–3180 Search PubMed;
(b) E. E. Dueno, T. Ray, R. N. Salvatore, C. Zambrano, M. Zellar and A. D. Hunter, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2007, 63, 3533–3534 Search PubMed.
- C. Zambrano, J. Kass, E. Dueno, Y. Ke and H.-C. Zhou, J. Chem. Crystallogr., 2006, 36, 67–70 CrossRef CAS.
- J. Han, X. Song, L. Liu and C. Yan, J. Inclusion Phenom. Macrocyclic Chem., 2007, 59, 257–263 CrossRef CAS.
- T. Gerkensmeier, C. Agena, W. Iwanek, R. Frohlich, S. Kotila, C. Nather and J. Mattay, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 1063–1973 CAS.
-
(a) M. M. Garcia, C. V. Launizar, F. L. Ochoa, A. Toscano, G. J. Chen and R. Cruz-Almanza, Supramol. Chem., 2000, 11, 255–261 CrossRef CAS;
(b) L. Zhaoa, Y. Wan, S. Huang, G. Liu, L. Chen, S. Yue, W. Zhang and H. Wu, Lett. Org. Chem., 2013, 10, 298–301 CrossRef;
(c) S. F. Alshahateet, S. A. Al-Trawneh, W. A. Al-Zereini and S. S. Al-Sarhan, Jordan J. Chem., 2014, 9, 170–186 Search PubMed.
- S. F. Alshahateet, F. Kooli, M. Messali, Z. M. A. Judeh and A. S. ElDouhaibi, Mol. Cryst. Liq. Cryst., 2007, 474, 89–110 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information regarding experimental procedures is available. CCDC 1405215–1405227. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01792k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a