
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highlights from Faraday Discussion: Designing New Heterogeneous Catalysts, London, UK, April 2016
Nico
Fischer
*a,
Haresh G.
Manyar
*b and
Alberto
Roldan
*c
aInstitute for Catalysis Research and DST-NRF Centre of Excellence in Catalysis c*change, Department of Chemical Engineering, University of Cape Town, 7701, Cape Town, South Africa. E-mail: nico.fischer@uct.ac.za
bCenTACat, School of Chemistry and Chemical Engineering, Queen's University, Belfast, David Keir Building, Stranmillis Road, Belfast BT9 5AG, UK. E-mail: h.manyar@qub.ac.uk
cSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: roldammartineza@cardiff.ac.uk
First published on 16th June 2016
Abstract
The Faraday Discussion on the design of new heterogeneous catalysts took place from 4–6 April 2016 in London, United Kingdom. It brought together world leading scientists actively involved in the synthesis, characterisation, modelling and testing of solid catalysts, attracting more than one hundred delegates from a broad spectrum of backgrounds and experience levels – academic and industrial researchers, experimentalists and theoreticians, and students. The meeting was a reflection of how big of an impact the ability to control and design catalysts with specific properties for particular processes can potentially have on the chemical industry, environment, economy and society as a whole. In the following, we give an overview of the topics covered during this meeting and briefly highlight the content of each presentation.
The first experience of a Faraday Discussion is always an eye-opener about how lively conference discussions can become. Indeed, these meetings focus genuinely on in-depth and fruitful discussions rather than an exposition of results with limited interaction between researchers. The format dictates that presenters submit full papers several months in advance which are lightly reviewed before all the papers are circulated, both electronically and in hard copy, to every delegate. Participants arrive at the conference prepared with their comments and queries after studying the manuscripts. At the conference, after a plenary Opening Lecture, the authors, in groups of three to four, present their work for five minutes, each followed by extensive discussion of 20–25 minutes for each paper. After this intense peer review for each of the papers presented, the session is opened to debate where general questions and comments are exposed to the panel of speakers, and in fact anyone else in the audience. All thoughts and considerations form part of the peer review process and are sequentially recorded and published in conjunction with the accepted manuscripts. This practice of a discussion-review with participants of different levels in the relevant field is both a daunting and a very enriching experience at the same time. This has been the trend since the first Faraday Discussion debated “osmotic pressure” in 1907 in London, which clearly sets it apart from any other conference to date.
The conference started on the 4th of April and once more London welcomed the Faraday Discussions attendees with blue skies and warm temperatures, contrasting the English meteorological stereotype. Although the discussion was held formally in The Geological Society, the social and poster sessions took place in the beautiful Palladian-style Burlington House, residence of the Royal Society of Chemistry (RSC). The conference began with a welcoming note from the conference chair Graham Hutchings (Cardiff University) who also headed the scientific committee constituting Michael Bowker (Cardiff University), Robbie Burch (Queen's University Belfast), Philip Landon (Sasol Technology (UK) Ltd), Jacob Moulijn (Delft University of Technology) and David Willock (Cardiff University).
Opening lecture
The conference was opened by Avelino Corma (Instituto de Tecnología Química, Spain), who delivered an exciting introductory lecture highlighting the importance of catalyst design for the success of the chemical process industry and gave an overview of the historic approach to basic catalyst design using first generation tools and how they have now evolved into advanced techniques guided by the mechanistic knowledge obtained from surface characterization, in situ spectroscopy and molecular modelling (DOI: 10.1039/C6FD00066E). Prof. Corma put emphasis on how we can design better catalysts by following the methodology based on the mechanistic knowledge of the reaction to be catalysed, wherein catalyst synthesis is followed using structural characterization and molecular modelling to correlate the structure of the active sites with the catalytic activity. He demonstrated this methodology by way of an example using well-defined gold nanoparticles in the selective hydrogenation of nitroaromatics. Prof. Corma also presented the design of a naked metal cluster as a catalyst using the example of sub-nanometric Cu(I) clusters of 2–7 atoms catalyzing a variety of cross-coupling reactions of iodoaromatics and iodoalkenes. In this way, Prof. Corma set the scene for the exciting Faraday Discussion on the design of heterogeneous catalysts. During the conference, Prof. Corma was awarded the prestigious Spiers Memorial Award 2016 for the introduction of new concepts for the synthesis of microporous and mesoporous inorganic materials and their application in heterogeneous catalysis which have resulted in an extensive range of industrial applications. The Spiers Memorial Award is presented once a year to the introductory lecturers who are likely to provide the most stimulating and wide-ranging introduction to the discussion.Session 1: Designing new catalysts: synthesis of new active structures
Following the opening lecture, the first session of the conference, chaired by Jacob Moulijn, was opened with a captivating presentation by Charles Campbell (University of Washington, USA) who presented the degrees-of-rate-control analysis of each adsorbed intermediate and transition state for the selective oxidation of n-butane to 1-butanol with O2 using a model for sterically constrained Ag3Pd(111) bimetallic nanoparticles encapsulated within a metal organic framework as a catalyst (DOI: 10.1039/C5FD00198F). The authors used a Ag3Pd(111) surface decorated with an immobile ring of helium atoms to resemble the pores present within the MOF-encapsulated metal nanoparticle catalyst (Fig. 1). The authors calculated the adsorption energies and kinetic parameters using DFT and developed the microkinetic model for the prediction of reaction rates and product selectivities in the activation of a C–H bond using n-butane oxidation as an example. The insights obtained from the degrees-of-rate-control analysis of the rate controlling species suggested the modification of the feed by addition of water, which not only increased both the reaction rate of the n-butane oxidation and selectivity to 1-butanol significantly but also decreased the O2 consumption. Following on from the simulation of MOF-mimetic catalyst pores, the session continued along the theme of new catalyst active structures with a second presentation given by Richard Palmer of the University of Birmingham, UK (DOI: 10.1039/C5FD00178A). The authors presented a relatively new cluster beam approach for model catalyst synthesis, typically by generating the metal atomic clusters in the gas phase and depositing onto the catalyst support as a beam in high vacuum. Using this approach, the authors synthesized Pd, PdSn and PdTi clusters supported on γ-Al2O3 and evaluated their catalytic activity in the selective hydrogenation of alkynes to alkenes. In the hydrogenation of 1-pentyne, the PdTi cluster catalysts showed high selectivity (85–90%) to 1-pentene at >95% 1-pentyne conversion. The authors also presented the similar superior performance of PdTi cluster catalysts for the selective hydrogenation of 3-hexyn-1-ol to cis-hexen-1-ol as compared vis-à-vis to conventional catalysts prepared via impregnation. The authors also described the modification of the cluster beam source design to scale-up the synthesis of cluster catalysts by several orders of magnitude. The third presentation, delivered by Robert Madix (Harvard University, USA), moved back to oxidation reactions using a recently reported new type of unsupported, nanoporous gold catalyst, which is shown to be highly active and selective in key oxidation processes, as well as being very robust under the reaction conditions. The author presented the insights obtained in the activation of molecular O2 on nanoporous gold (npAu) catalysts using a temporal analysis of products (TAP) reactor (DOI: 10.1039/C5FD00161G). Dr Madix presented the results under the TAP conditions showing low surface coverage of atomic oxygen on np(Ag)Au catalysts indicating that the active site density for O2 dissociation on a npAu catalyst was low, thus highlighting the significance of the AgAu bimetallic active site responsible for O2 activation. The results from this study indicated that the molecular O2 dissociation was associated with surface silver.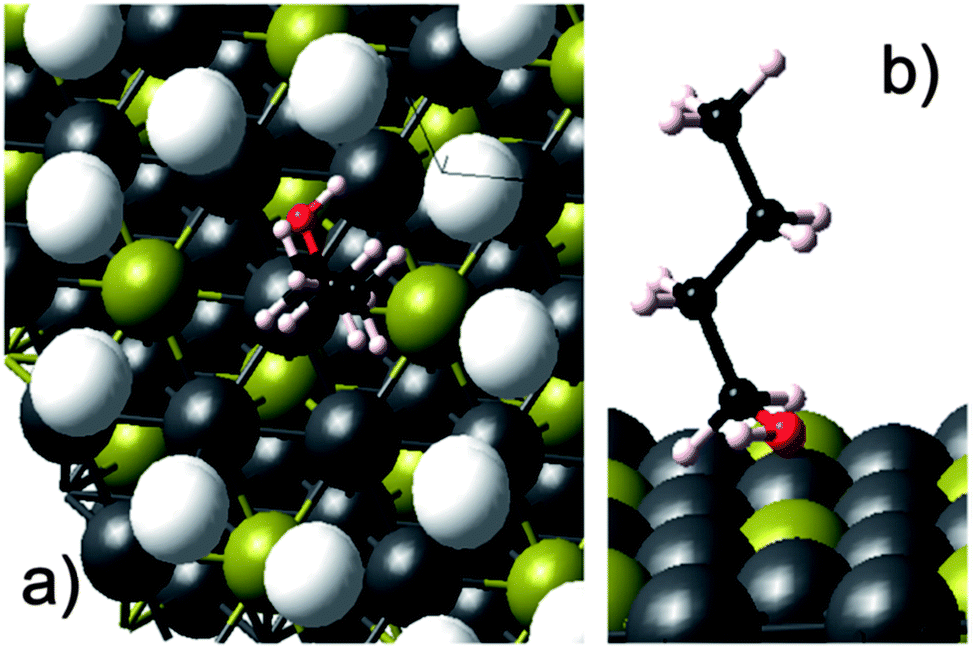 | ||
| Fig. 1 (a) Top view of adsorbed n-butanol on the He ring-encapsulated Ag3Pd(111) surface. (b) Side view of the same system shown in panel (a) but with the He atoms removed to view the surface sites better. Ag = grey, Pd = yellow, C = black, H = off-white, O = red, He rings = white (DOI: 10.1039/C5FD00198F). | ||
The second part of the first session was opened by Bert Weckhuysen (Utrecht University, The Netherlands), with a relatively simple but very exciting photo-spectroscopy technique for catalyst characterization using a standard DSLR photo-camera with a macro lens and FCC catalyst particles as exemplary catalysts (DOI: 10.1039/C5FD00210A). Prof. Weckhuysen used the images of the individual catalyst particles obtained after selective staining with substituted styrene molecules to link the observed colour changes with the catalyst physical properties such as age, micropore volume and catalytic cracking activity data. The authors explained that the colour of the stained samples was related to the type of zeolite and it could be used to identify and quantify the number of FCC-ZSM-5 and FCC-Y particles (Fig. 2). The next presentation was delivered by Wataru Ueda (Kanagawa University, Japan), who described the development of new crystalline complexes formed by the assembly of ε-Keggin polyoxomolybdate units with metal ion linkers in a diamond topology as well as the hexagonal polyoxomolybdate units stacked to form prismatic clusters as molecular nanowires (DOI: 10.1039/C6FD00006A). Prof. Ueda demonstrated that the material possesses good catalytic activity in O2 adsorption, selective oxidation of methacrolein and hydrolysis of cellobiose. The session further progressed with a presentation delivered by Annette Trunschke (Fritz-Haber-Institut der Max-Planck-Gesellschaft, Germany), who described the influence of catalyst structure and the balance of acid–base and redox sites on C–H activation using ethanol oxidation as the probe reaction and monoclinic as well as rod-shaped nano-crystalline manganese tungstate as catalysts (DOI: 10.1039/C5FD00191A). The authors proposed a mechanism of particle growth by dissolution and re-crystallisation that led to an increased aspect ratio which in turn affected the product distribution. The last presentation of the session was delivered by Peter Wells (UK Catalysis Hub, Research complex at Harwell; University College London, UK), who presented the design and stabilisation of high surface area iron molybdate for the selective oxidation of methanol to formaldehyde (DOI: 10.1039/C5FD00153F). The authors successfully stabilized high surface area core–shell type MoOx@Fe2O3 catalysts based on the 5-fold increase in the surface area of the haematite support resulting from doping 5 wt% aluminium into the core and achieved superior catalytic activity in methanol oxidation with a high yield (94%) of formaldehyde.
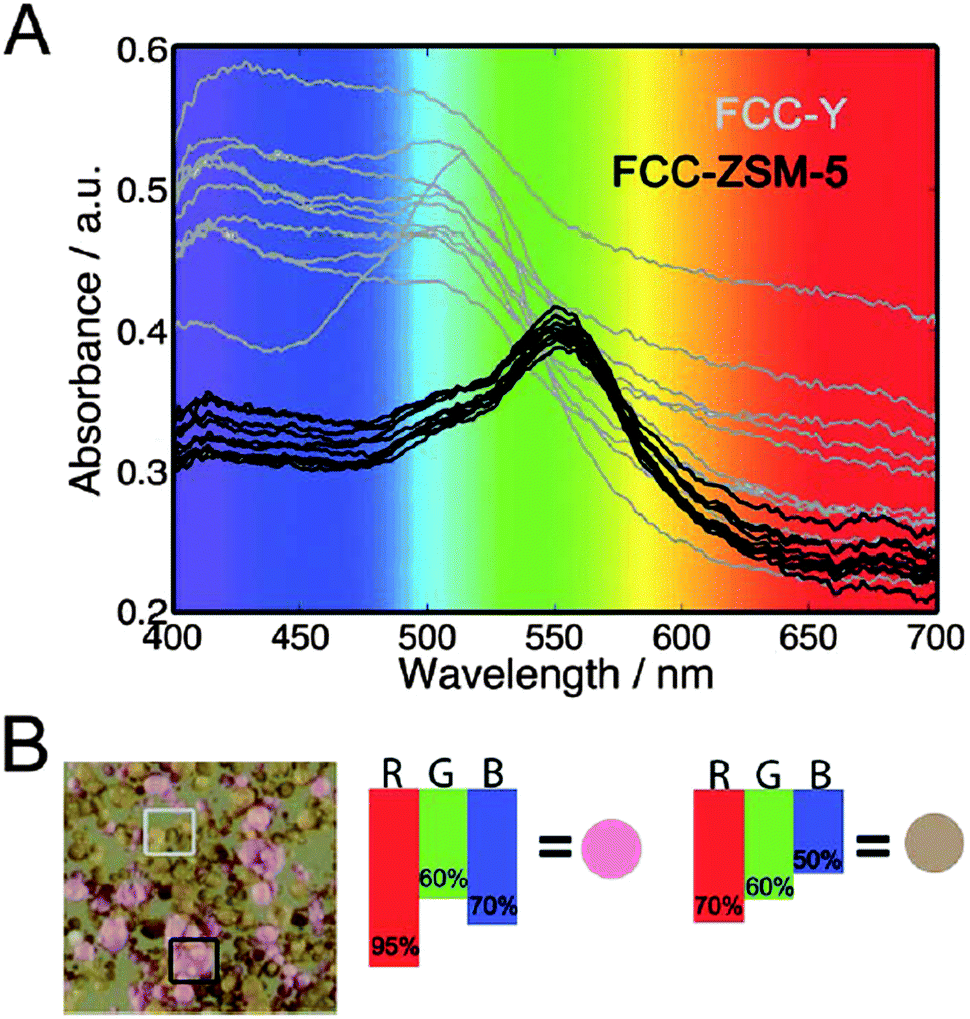 | ||
| Fig. 2 Plots illustrating the relationships between zeolite type, colour and specific absorption maxima (DOI: 10.1039/C5FD00210A). (A) Absorption spectra of samples FCC-ZSM-5 (black) and FCC-Y (gray); (B) photograph of a mixture of samples FCC-ZSM-5 (pink) and FCC-Y (orange). The transmission percentages for red, green, and blue are displayed with the resulting color, explaining the spectroscopic base on which a distinction can be made between FCCZSM-5 and FCC-Y. | ||
As per the tradition of Faraday Discussions, each short 5 minute presentation was followed first by an individual thorough and detailed discussion on the results presented by the respective authors and then with the combined discussion open for all to comment on the advancement of new active structures of solid catalysts, the controlled design of active sites and the evolution of catalyst synthesis techniques. The first session was followed by a lightning session of rapid flash presentations selected by the organizers to highlight selected poster contributions.
Session 2: Catalyst design from theory to practice
The second day started with the Catalyst design from theory to practice session chaired by David Willock. This session focused on computational tools to simulate catalytic processes. The first contribution described the catalytic activity of an iron sulphide spinel structure in the reduction of carbon dioxide (DOI: 10.1039/C5FD00186B). The author carried out the study using density functional theory (DFT) including dispersion interactions and an electron correlation correction. The results and discussion of this paper focused on one surface of the catalyst and explored the reaction pathway network with more than 200 reaction intermediates on all the different active sites. This intensive investigation concluded with the formation of formic acid and methanol on particular surface sites under mild conditions, see Fig. 3. The kinetic model showed the rate limiting steps whose identification is indispensable to improve the conversion and yields. In the discussion of the paper during the peer review process questions were raised about the dynamism of the surface, and the source of the hydrogen atoms as well as the comparison with an enzymatic catalyst, which inspired the project. The following 5 minute talk was by Maganas from the Max Planck Institute for Chemical Energy Conversion, who used advanced quantum chemical calculations to address spectroscopic simulations in the study of heterogeneous and homogeneous catalysts (DOI: 10.1039/C5FD00193E). Particularly, they employed restricted open shell configuration interactions and the time-dependent theory of electronic spectroscopy. They combined these techniques to simulate the X-ray absorption measurements and resonance Raman spectroscopy of two related systems: silica-supported vanadium oxide VOx/SBA-15 and K[VO(O2)Hheida] domains. The audience formulated several comments on the results and methods employed during the spectra simulation bringing new ideas to the simulation of spectra. The last presentation before the discussion had important implications for one of the holy-grails of our times: H2 evolution from water. Plaisance (TU München) presented computational results on the evolution of molecular oxygen from a cobalt oxide catalyst (DOI: 10.1039/C5FD00213C). This paper included the catalytic cycle describing the electronic structure of the reaction intermediates, clarifying the electron transfer mechanisms, vital for designing an optimal catalyst. The analysis of the reaction kinetics revealed the activity trends for water addition as a function of oxidation potentials at different overpotentials. Observations on the reliability of this methodology and its accuracy on reaction energies were raised. According to the Faraday Discussions format, individual comments and queries were followed by a combined and detailed discussion on the applicability of computational tools and their disadvantages when the models are compared with real catalyst experiments. Subjects such as heterogeneous and solid catalysts, dynamical surface processes, and reactivity on defects were mentioned and discussed in detail.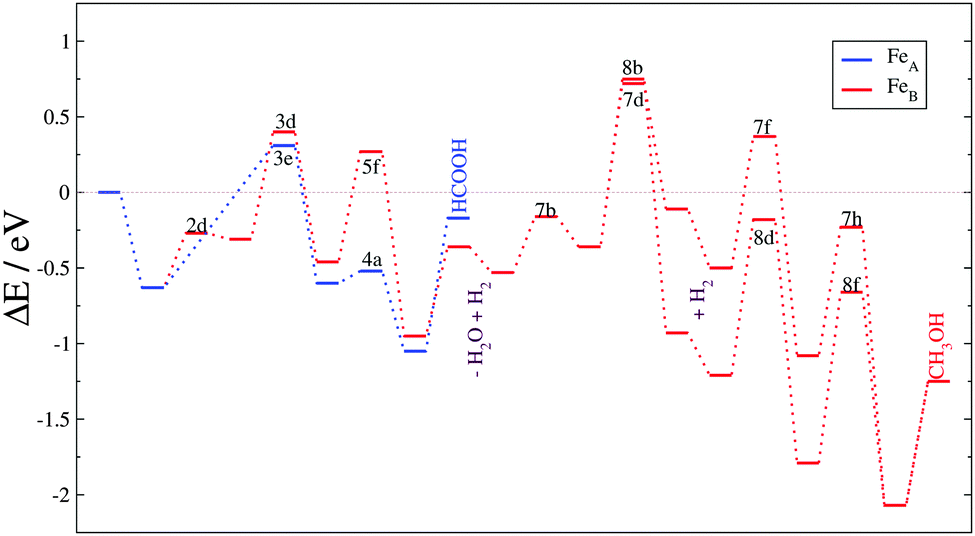 | ||
| Fig. 3 Reaction energy profiles for the CO2 hydrogenation process on a Fe3S4{111} surface (DOI: 10.1039/C5FD00186B). | ||
The second part of the session started after a morning tea break where many posters caught the attention of the audience. Professor Sauer opened the round of presentations with his interesting research on bulky and thin film zeolitic materials carried out using periodic DFT and quantum mechanics combined with molecular mechanics (QM/MM) (DOI: 10.1039/C5FD00207A). Interestingly it could be shown that the deprotonation of ammonia and pyridine on Brønsted sites is more favourable on two-dimensional sites due to the smaller effective dielectric constant of these materials. The five minute talk passed quickly and O'Malley stepped on stage to introduce part of his work on zeolites carried out at University College London (DOI: 10.1039/C6FD00010J). In this case, however, the modelling of the acid sites and the reaction mechanism was shown. The method used to carry out this research was QM/MM. They explored both H-ZSM-5 and H–Y zeolite structures in the context of methanol conversion to hydrocarbons and olefins (Fig. 4). They explored the energies of adsorption and reaction on Brønsted acid sites and found correlations between species geometries and energies. The final speaker of the second session presented the latest results on the oxygen reduction reaction on nanoparticles of gold and palladium (DOI: 10.1039/c5fd00233h). The persistence of this topic indicates how important and challenging it is to design an effective catalyst for this process which is the reverse step of obtaining molecular hydrogen from water. An alloy catalyst was suggested and peroxo species on the surface were identified as intermediates in the electrocatalytic reduction of water peroxide. The structure of the metal alloy was an issue of discussion following the previous debate on surface dynamics (Fig. 5). In situations like that, it is reasonable to remember that computational models are just models trying to mimic the actual catalyst. A model based on an individual metal as a catalytic site under conditions of temperature and in the presence of surface and solvent species, perhaps does not represent the dynamics of the alloy surface but it may be a situation where the surface may present this configuration. It is then responsibility of modelers to analyse the relative stability of the catalytic centres to ensure the simulated situation will represent the main case under experimental conditions. The general consensus was to consider, once more, that computational methods are useful tools to shed light on the determination of reaction pathways and general trends. The in silico experiments, however, need to close the gap between models and actual experiments, perhaps improving models, broadening them to catalytic conditions and definitely exploring non-ideal representations of the surfaces.
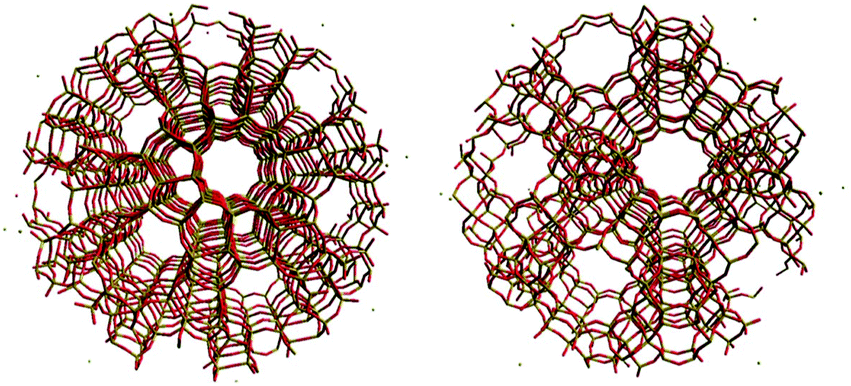 | ||
| Fig. 4 Spherical embedded cluster models of the I2-T12 centred H-ZSM-5 system (left) and the zeolite Y system (right) (DOI: 10.1039/C6FD00010J). | ||
 | ||
| Fig. 5 STM images of Ag on 2 ML MgO(100)/Ag(100) annealed at different temperatures (a–d) (DOI: 10.1039/c5fd00143a). | ||
Session 3: Bridging model and real catalysts
Professor Freund from the Fritz-Haber-Institute of the Max-Plank-Gesellschaft in Berlin started the third session chaired by Mike Bowker. His report (DOI: 10.1039/C5FD00143A), focusing on the activation of CO2 on gold particles supported on a 1 ML MgO(100)/Ag(100) surface and the hydrogenation of acrolein over Pd on a Fe3O4/Pt(111) substrate, illustrated that both the metal support interface, as well as present spectator species can have a significant impact on the activity and selectivity of a catalyst even resulting in different reaction mechanisms. The annealing of the Au particles at higher temperatures changed their morphology from flat islands supporting charge transfer from the underlying Ag substrate to three dimensional Au particles lacking the electron donation from the support and consequently not yielding (CO2)2− dimers and subsequently absorbed oxalate (Fig. 5). On larger Pd crystallites exhibiting Pd(111) planes an over layer of oxopropyl species directs the adsorption of acrolein and guides the C–O hydrogenation over the C–C double bond hydrogenation. If Pd(111) surfaces are not exposed, such as in small crystallites, Pd favourably hydrogenates the double bond yielding propanal. Triangular planar MoS2 nanoparticles on Au(111) with different sizes and edge terminations (different levels of sulphidation as well as S vs. Mo termination) were studied theoretically by Bruix (DOI: 10.1039/C5FD00203F). Simulated STM images showed the metallic character of the edge sites independently of termination. Reducing the sulphidation of Mo terminated particles resulted in a loss of the metallic characteristics which was not so in the S terminated case. The modelling of H and S adsorption further indicated that clear differences exist between the different available sites (edge, corner, near-edge and basal plane). The third presentation by Gross of the Hebrew University of Jerusalem (DOI: 10.1039/C5FD00194C) introduced a TR-FTIR nano-spectroscopy technique to study the chemical properties of adsorbates with high spatial resolution. As a sample system the interaction of OH functionalized N-heterocyclic carbenes on a Pt surface was chosen. Only under harsh oxidizing conditions could the detachment of ligands be observed. Generally the method allows the study of molecular behaviour on single nanoparticles. The presentations resulted in a discussion on the possibility of studying real and dynamic catalytic surfaces and their difference to model systems studied under UHV conditions. This theme would resurface in multiple exchanges during the progress of the meeting.Cynthia Friend from Harvard University restarted the session by reporting on the relationship between the relative binding strength of different substrates on a catalytic surface with the observed catalytic performance (DOI: 10.1039/C5FD00192G). Using displacement reactions a hierarchy for the binding strength was established experimentally on an Au(111) surface, see Fig. 6. In homologous series such as alcohols the weak van der Waals forces that increase in strength with increasing chain length start to play a significant role in the binding energy and therefore in the competition for adsorption sites. Fluorinated functional groups decrease said energy via Pauli repulsion with the surface. The effect of the zeolite binder/kaoline interaction, as is present in industrial catalysts for processes such as fluid catalytic cracking and the conversion of methanol to hydrocarbons, was presented by Whiting (DOI: 10.1039/C5FD00200A). The acid catalysed oligomerization of thiophene was chosen as a staining probe reaction and the progress was monitored through in situ UV-vis spectroscopy and confocal fluorescence microscopy. In the presence of kaoline the activity of the ZSM-5 was drastically reduced which was attributed to the formation of a hard shell upon heat treatment. In the presence of Al2O3 as a binder, aluminium migration was observed yielding a higher activity towards the ring opening of thiophene. SiO2 in turn enhances the formation of larger oligomers through a direct binder–substrate interaction. The final presentation of the session was given by Emma Gibson from the UK Catalysis Hub in Harwell/Cardiff University (DOI: 10.1039/C5FD00225G). Hematite particles as well as hematite particles with a layer of MoOx were tested in the selective oxidation of methanol to formaldehyde. While Fe2O3 oxidizes adsorbed methanol via a formate species to CO2, the presence of a closed layer of Mo blocks this pathway and yields formaldehyde with a high selectivity.
 | ||
| Fig. 6 Hierarchy of the relative binding affinities of various molecules on Au(111) (DOI: 10.1039/C5FD00192G). | ||
Session 4: Application of novel catalysts
The meeting culminated in the last session with reports on the application of novel catalysts. Using a supercritical anti-solvent process lanthanum based perovskites were prepared by varying the transition metal in the B site. These high surface area materials could be applied as support materials for AuPt catalysed oxidation. Kondrat and co-workers (DOI: 10.1039/C5FD00187K) reported that by changing the metal site, the reaction mechanism could be influenced ranging from a sequential oxidation pathway yielding glyceric/tartronic acid to an initial oxidation and dehydrogenation pathway which results in the formation of lactic acid with high selectivity (Fig. 7). Haresh Manyar presented work conducted at the Queen's University in Belfast on porous manganese oxide (OMS-2) for the hydrogenation of halogenated arenes (DOI: 10.1039/C5FD00227C). It was reported that OMS-2 resulted in no dehalogenation. Addition of Pt increased the reaction rate without changing the reaction mechanism, i.e. a similar selectivity was achieved. However if Pt was replaced by Pd, significant dehalogenation of the substrate was observed. The authors conclude that the reaction proceeds on the surface of OMS-2 in the presence of Pt and that Pt solely acts as hydrogen dissociation centre. In the presence of Pd, the reaction proceeds on the metal surface resulting in a different product distribution. These conclusions were supported by hydrogenation experiments of nitrobenzene. Based on a previously proposed mechanism for the production of DME from methanol over η-alumina the group of Lennon (DOI: 10.1039/C5FD00202H) proposed and tested a reaction mechanism for the formation of methyl chloride over the same acid catalyst. It could be shown that the doping of alumina with group one metal salts, and specifically CsCl, did result in a decrease in the rate of formation of methyl chloride but in turn significantly increased the selectivity of the catalyst through ‘capping’ of the strong and medium strength acid sites. In the industrial environment the economic advantages of the increase in selectivity clearly surpass the loss in formation rate. The final paper before tea was presented by Henrik Svengren from Stockholm University (DOI: 10.1039/C5FD00169B). The use of a (Co,Ni,Mn)3Sb4O6F6 solid solution as an electrode material in the water oxidation reaction was reported. Compared to a CoOx based electrode the oxofluoride materials showed an improved activity and also stability. While SEM images showed a morphological change after 0.5 hours (without further changes up to 64.5 hours) under reaction conditions, no compositional changes could be measured.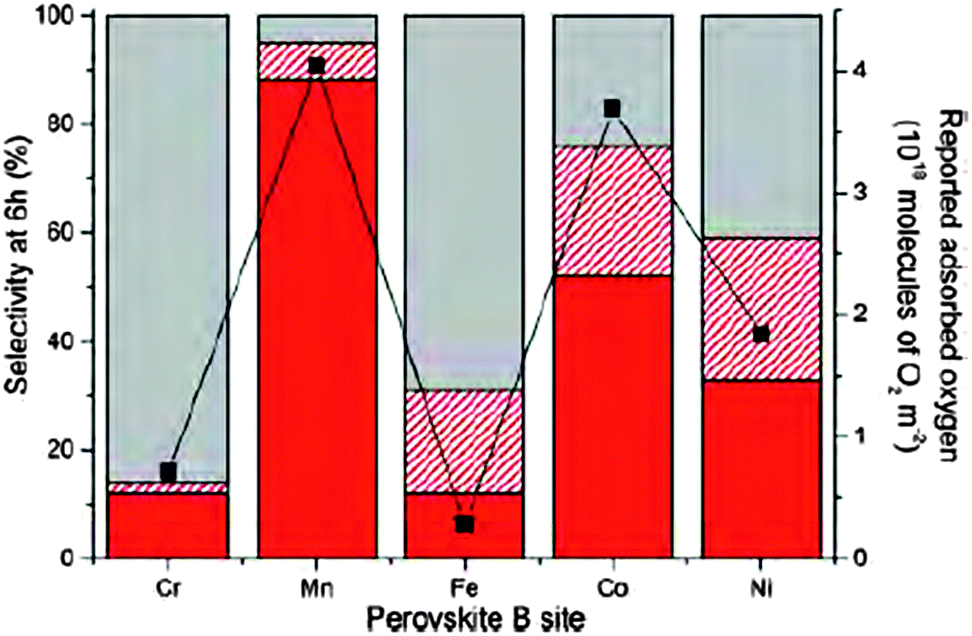 | ||
| Fig. 7 Selectivity profiles of AuPt/LaBO3 of C3 oxidation products (red), C–C scission products (dashed red) and lactic acid (grey), compared to reported oxygen adsorption (black squares and right y-axis) (DOI: 10.1039/C5FD00187K). | ||
The last set of papers was commenced with a study of Au core Pd shell catalysts for the hydrogenation of acetylene to ethene (DOI: 10.1039/C5FD00188A). The major challenge in this industrially relevant process is the suppression of the full hydrogenation to ethane. Different Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au atomic ratios (0.2, 1.5 and 3.3) were realized by addition of a Pd solution to pre-formed Au nanoparticles (Fig. 8). The lowest concentration of Pd resulted in an incomplete Pd shell around the Au particles without the formation of Pd monometallic crystallites. The isolated Pd sites in conjunction with the free Au sites resulted in a high selectivity to ethene and oligomers while very little total hydrogenation was observed. Higher concentrations of Pd result in a complete shell in addition to Pd monometallic nanoparticles. These particles behave similarly to pure Pd catalysts with over 50% selectivity to ethane. A potential catalyst to unlock the stored hydrogen from ammonia was presented by Hunter (DOI: 10.1039/C5FD00179J). Lithium–calcium imide is studied as a potential candidate due to its increased stability and solid form under the reaction conditions compared to previously studied materials such as the sodium/sodium amide system or non-stoichiometric lithium imide. The studied material turned out to be the best performing ammonia decomposition catalyst from this family of materials reported to date. In the last paper of the meeting, Landau from the Ben-Gurion University in Negev, Israel, reported on the use of various iron containing oxide materials as an Fe matrix for CO2 hydrogenation via the reverse water gas shift reaction followed by a Fischer–Tropsch like reaction (DOI: 10.1039/C5FD00174A). The iron in all phases was converted under reaction conditions to a mixture of iron carbides and oxides in different compositions, directed by the initial oxide matrix. Fe–Al–O spinel and Fe–Ba–hexaaluminate precursors yielded the highest selectivity towards long chained hydrocarbons.
:Au atomic ratios (0.2, 1.5 and 3.3) were realized by addition of a Pd solution to pre-formed Au nanoparticles (Fig. 8). The lowest concentration of Pd resulted in an incomplete Pd shell around the Au particles without the formation of Pd monometallic crystallites. The isolated Pd sites in conjunction with the free Au sites resulted in a high selectivity to ethene and oligomers while very little total hydrogenation was observed. Higher concentrations of Pd result in a complete shell in addition to Pd monometallic nanoparticles. These particles behave similarly to pure Pd catalysts with over 50% selectivity to ethane. A potential catalyst to unlock the stored hydrogen from ammonia was presented by Hunter (DOI: 10.1039/C5FD00179J). Lithium–calcium imide is studied as a potential candidate due to its increased stability and solid form under the reaction conditions compared to previously studied materials such as the sodium/sodium amide system or non-stoichiometric lithium imide. The studied material turned out to be the best performing ammonia decomposition catalyst from this family of materials reported to date. In the last paper of the meeting, Landau from the Ben-Gurion University in Negev, Israel, reported on the use of various iron containing oxide materials as an Fe matrix for CO2 hydrogenation via the reverse water gas shift reaction followed by a Fischer–Tropsch like reaction (DOI: 10.1039/C5FD00174A). The iron in all phases was converted under reaction conditions to a mixture of iron carbides and oxides in different compositions, directed by the initial oxide matrix. Fe–Al–O spinel and Fe–Ba–hexaaluminate precursors yielded the highest selectivity towards long chained hydrocarbons.
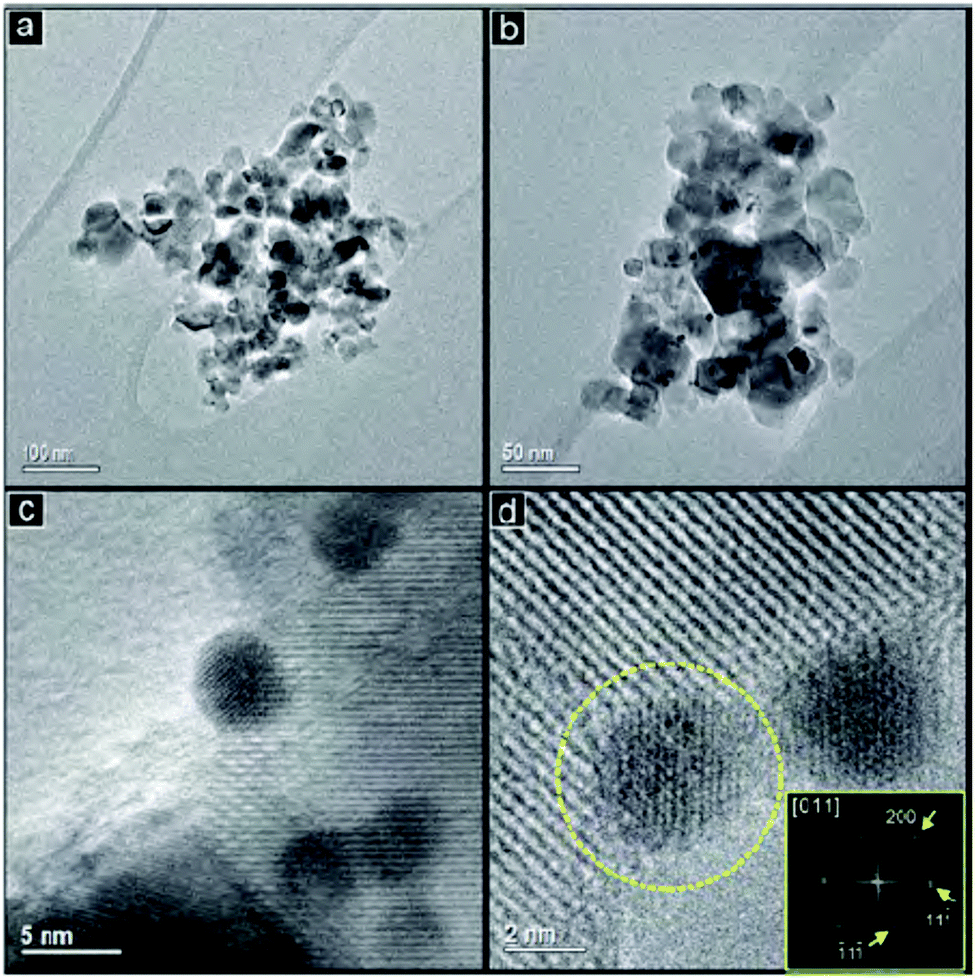 | ||
| Fig. 8 TEM images with different magnifications (a–d) of Pd/Au gold nanoparticles with an atomic ratio Pd:Au of 3.3 (DOI: 10.1039/C5FD00188A). | ||
Closing lecture/concluding remarks
The concluding remarks were presented by Prof. Bruce Gates (UC Davis, USA) (DOI: 10.1039/C6FD00134C). He recapitulated the meeting by giving a brief overview of all the topics covered during the Faraday Discussion but also highlighting that the field of heterogeneous catalysis is even wider and certain areas did not feature. He attempted to outline several questions and potential directions for the future development of catalyst design, focusing on the interaction of experimental and theoretical work as well as the general struggle to identify the ‘real’ working catalyst surface and active site in industrially relevant heterogeneous systems. Importantly, he pointed out that in spite of the broad range of topics covered in the meeting, it did not exhaust the full potential. Some of the important topics in catalysis were not represented like those that include actual reaction kinetics, and the use of amorphous materials as catalysts and he emphasized the need to address the nature of these amorphous surface layers. It was generally agreed by the community that closer collaboration between industry and academia can help us to achieve faster catalyst optimization and scale up.This Faraday Discussion illustrated that the design of catalysts is a broader area which includes, besides conventional modes of organic and inorganic reactivity, physical properties of the materials, e.g. the interaction between catalyst and support, and sintering of the nano particles.
The concluding remarks were followed by well-deserved acknowledgements to the hosts and organisers, before one final lunch brought a very successful and enjoyable Faraday Discussion to a close. The delegates went their separate ways with plenty to think about following an intense but stimulating three days of discussion.
| This journal is © The Royal Society of Chemistry 2016 |