
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitation in the regioselectivity of acylation of glycosyl diglycerides: total synthesis of a Streptococcus pneumoniae α-glucosyl diglyceride†
Mark B.
Richardson
*,
Dylan G. M.
Smith
and
Spencer J.
Williams
 *
*
School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: sjwill@unimelb.edu.au
First published on 22nd December 2016
Abstract
The fidelity of acylation regioselectivity in the synthesis of mixed glycosyl diacylglycerols can be accurately measured by quantitative 13C NMR spectroscopy using a 1-13C-labelled fatty acid and a paramagnetic relaxation enhancement agent. Exquisite regioselectivity is achieved using a stepwise acylation/substitution of a glycosyl β-bromohydrin, which is applied to the total synthesis of Streptococcus pneumoniae Glc-DAG-s2.
Glycosyl diacylglycerols (glycosyl diglycerides) are a diverse group of glycolipids produced by terrestrial and marine plants and algae, bacteria, and archaea.1 The sugar residue is typically found at the primary position (usually sn-3), and distinct fatty acyl groups located at the sn-1 and sn-2 positions. The arrangement of acyl groups is critical for immune recognition of glycosyl diglycerides. Glycosyl diglycerides bind to the antigen-presenting molecule CD1d, and the resulting CD1d–glycolipid complex is displayed on the cell surface of antigen presenting cells where the complex can be recognized by T cell receptors (TCR) on invariant natural killer T (iNKT) cells.2 iNKT cells are stimulated by Streptococcus pneumoniae Glc-DAG-s2 α-glucosyl diglyceride (1), bearing a palmitoyl group at the sn-1 position, and a cis-vaccenoyl group at the sn-2 position (sn-1/sn-2, C16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0/C18:1-cis-Δ11), but not the C18:1-cis-Δ11/C16:0 regioisomer.3 Similar observations extend to Borrelia burgdorferi BbGL-IIc α-galactosyl diglyceride (C18:1/C16:0), but not the C16:0/C18:1 regioisomer;4 and Mycobacterium smegmatis Gl-A α-glucuronosyl diglyceride (C19:0/C16:0; where C19:0 is R-tuberculostearic acid), but not the (C16:0/C19:0) regioisomer.5 The structural basis for sensitivity to the acyl group arrangement is well understood. CD1d has two lipid-chain binding pockets (A′ and F′) and glycosyl diglycerides bind with their lipids within these pockets and the carbohydrate head-group projecting at the surface of CD1d for TCR binding. In the case of the B. burgdorferi glycolipids, X-ray structures revealed preferential binding for oleoyl (C18:0) groups in the A′ pocket irrespective of their location at the sn-1 or sn-2 positions, leading to different orientations of the galactosyl moiety and distinct structures of the glycolipid–CD1d antigen complex.6
:0/C18:1-cis-Δ11), but not the C18:1-cis-Δ11/C16:0 regioisomer.3 Similar observations extend to Borrelia burgdorferi BbGL-IIc α-galactosyl diglyceride (C18:1/C16:0), but not the C16:0/C18:1 regioisomer;4 and Mycobacterium smegmatis Gl-A α-glucuronosyl diglyceride (C19:0/C16:0; where C19:0 is R-tuberculostearic acid), but not the (C16:0/C19:0) regioisomer.5 The structural basis for sensitivity to the acyl group arrangement is well understood. CD1d has two lipid-chain binding pockets (A′ and F′) and glycosyl diglycerides bind with their lipids within these pockets and the carbohydrate head-group projecting at the surface of CD1d for TCR binding. In the case of the B. burgdorferi glycolipids, X-ray structures revealed preferential binding for oleoyl (C18:0) groups in the A′ pocket irrespective of their location at the sn-1 or sn-2 positions, leading to different orientations of the galactosyl moiety and distinct structures of the glycolipid–CD1d antigen complex.6
Owing to their trace concentrations and heterogeneity from natural sources, and frequent ambiguities in structure, chemical synthesis has been central to defining the structures of glycoglycerolipids, overcoming limited supply, and providing homogeneous samples of known structure for biomedical research. A critical challenge in the chemical synthesis of these compounds is ensuring that the glycerol is regioselectively acylated with high fidelity. Installation of two different acyl groups is typically performed in a stepwise manner through a β-hydroxy-ester intermediate in which acyl group migration is facile,7 occurring under both acidic and basic conditions, and even during chromatography on silica gel or florasil (Fig. 1a).8,9 While methods have been described that are claimed to mitigate acyl migration, it is difficult to quantify the degree of migration and thus any erosion of regioselectivity owing to the similar chromatographic mobilities and essentially identical NMR characteristics of the regioisomers. Preformed 1,2-diacylglycerols have been used as glycosyl acceptors in glycosylation reactions, but as β-hydroxy-esters are also prone to acyl group migration; commercial 1,2-diacylglycerols have been reported to be contaminated with their regioisomers.10
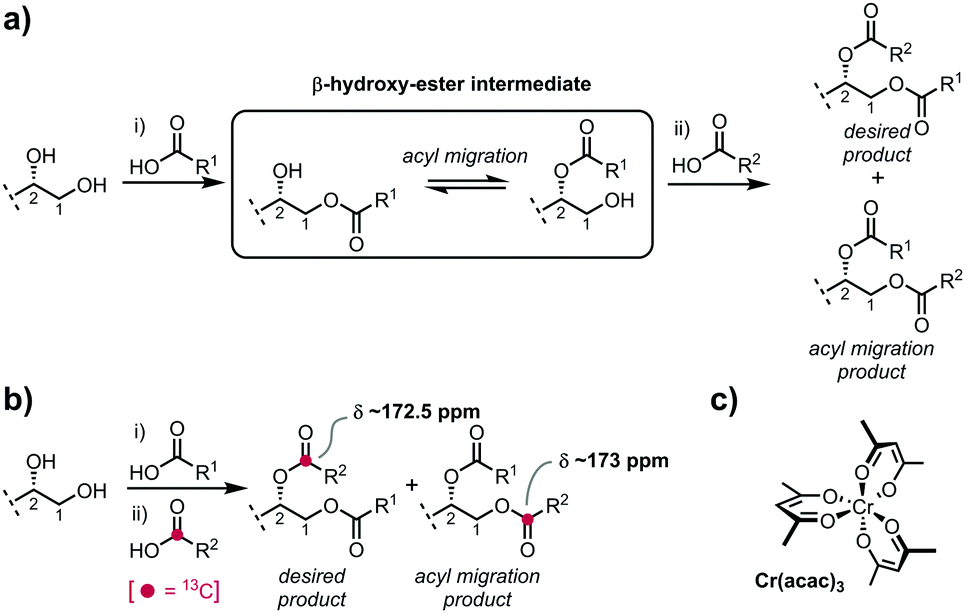 | ||
| Fig. 1 (a) Stepwise acylation can lead to mixtures of regioisomers through acyl migration of β-hydroxy-ester intermediates. (b) Monitoring regioselective acylation fidelity using a 13C-labelled fatty acid probe. (c) Cr(acac)3, a paramagnetic relaxation enhancement agent. | ||
Detection and quantification of acyl group migration is a challenging analytical problem. Fragmentation-mass spectrometry has been widely used for the structural characterization of acylation patterns,11,12 and relies upon qualitative differences in the rates of fragmentation of esters at the primary (sn-1) and secondary (sn-2) positions of a glycerol chain. While adequate for gross structural determination, it does not allow quantitation of low levels of regioisomers as significant fragmentation still occurs through the minor channel. Mazur et al. reported an NMR method for quantification of regioselectivity in the esterification of glycerol derivatives, which relies upon a subtle upfield shift of the ω − 3 13C atoms of primary esters of octanoic acid and decanoic acids, relative to the secondary esters, and allows measurement of the ratio of positional isomers to an accuracy of about 5%.13 The approach has not been applied to longer or more complex fatty acyl groups. Other studies have reported the use of NMR for the quantification of regioisomers of mixed glycerides containing long- and short-chain (acetyl, propionyl, butyryl) acyl groups that rely on distinctive chemical shift differences between the short and long chain groups,14 but cannot be used for glycerides bearing only long-chain acyl groups.
In the present work, we report an NMR method that allows accurate determination of the regioselective fidelity in acylation reactions. By inclusion of a 13C label into the carbonyl group of one of the fatty acyl groups, and use of a paramagnetic relaxation enhancement (PRE) agent with appropriate acquisition times, the abundance of the label at each position, (and hence the fidelity of regioselective acylation) can be measured by quantitative 13C NMR spectroscopy.
In previous work, we prepared a range of glycosyl diglycerides including β-gentiobiosyl diglycerides,15 α-glucosyl diglycerides,16 and α-glucuronosyl diglycerides.11 We consistently observed a chemical shift difference for the carbonyl group of an ester attached as a primary ester at the glycerol sn-1 position (δ ∼ 173 ppm), versus a secondary ester at the sn-2 position (δ ∼ 172.5 ppm), which was independent of the length of or functionalization within the fatty acyl chain. We reasoned that incorporation of a 13C label into the carbonyl group of one fatty acyl group would enable us to track whether this acyl chain was incorporated into the sn-1 or sn-2 positions. While 13C NMR is usually not considered to be quantitative, owing to differences in spin–lattice relaxation times (T1) of nuclei, compounded by very long relaxation times (>10 s) for quaternary carbons, paramagnetic relaxation enhancement (PRE) agents can dramatically reduce T1 times of quaternary carbons, allowing quantitative 13C NMR analysis.17 We focussed on Cr(acac)3, a non-shifting PRE agent with a high solubility in organic solvents.18 Unlike lanthanide shift reagents, Cr(acac)3 exhibits no angular dependence of its electron-nuclear dipole–dipole relaxation interaction.
Using a representative glycosyl diglyceride from previous work (1,2-di-O-[13-methyltetradecanoyl]-sn-glyceryl-2,3,4,6-tetra-O-benzyl-β-D-glucopyranoside)15T1 relaxation times of the ester carbonyls were estimated using the sign-inversion recovery method.17 In the absence of PRE agent, carbonyl 13C T1 relaxation times of around 15 s were observed; however, in samples containing 0.05 M Cr(acac)3T1 relaxation times were reduced to 0.5–1.0 s, comparable to published values.18 In order to reduce the potential for complicating nOe distortion an inverse-gated proton-decoupled experiment was employed in which the proton decoupler was switched off during the relaxation delay, and turned on only during data acquisition.17 Excellent results were obtained with a 10 s relaxation delay and a minimum of 2000 scans, which gave carbon resonances that could be integrated to <1% error across a spectrum.
For regioselective acylation experiments, commercial 1-13C-palmitic acid was employed. The atom% 13C labelling at C1 was determined to be 99.4% using negative ion electrospray ionization mass spectrometry, assuming that the remaining positions had isotope composition at natural abundance. The percentage ratio of regioisomers (R) can be calculated from the individual integrations of 13C resonances for carbonyl resonances at δ x ppm (labelled position) and δ y ppm (unlabelled position) using an acid of L13C atom% labelling, where N is the natural 13C abundance, eqn (1):
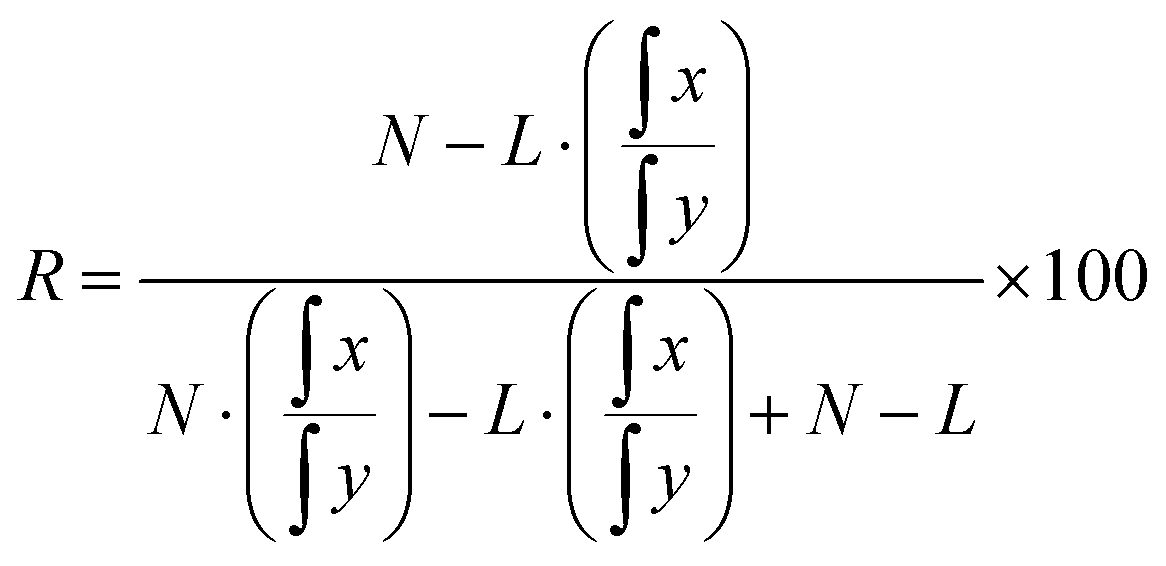 | (1) |
We first investigated the application of the method to the non-selective acylation of the diol 215 with a 1:1 mixture of commercial 1-13C-palmitic acid (1-13C–C16:0) and 10-methyldodecanoic acid (iso-C13:0) (Fig. 2a). This reaction should exhibit no regioselectivity, and assuming no significant differences in kinetic isotope effects for reactions at either of the sn-1 and sn-2 positions, an equimolar ratio of four compounds should be obtained, namely: double 1-13C-palmitic acid-incorporation (3), indiscriminate incorporation of 1-13C-palmitic acid and 10-methyldodecanoic acid at the sn-1 and sn-2 positions (4/5), and double 10-methyldodecanoic acid incorporation (6), leading to equal amounts of 13C label incorporation at each site. In accordance with this expectation, the resulting mixture of compounds displayed 13C NMR signals for the sn-1 and sn-2 carbonyls of equal intensity, indicating no regioselectivity.
We next explored a standard regioselective acylation utilizing stepwise Steglich esterifications of a glycosyl glycerol (Fig. 2b).10 Compound 2 was treated with 1.05 eq. of 10-methyldodecanoic acid, DMAP and 1.05 eq. DCC in CH2Cl2 at 0 °C for 12 h. To remove trace amounts of unreacted diol and residual acid, the monoglyceride was purified by flash chromatography. To limit the potential for acyl migration, purification was achieved in <15 min using chilled solvents and silica gel such that the temperature never exceed 10 °C at any point. Fractions were evaporated in vacuo and then immediately treated with 1.2 eq. of 1-13C-palmitic acid, DMAP and 1.2 eq. DCC in CH2Cl2 at room temperature, to afford after purification, the diester. Analysis of the diester revealed a 10:1 ratio of 13C carbonyl signals, corresponding to R = 92% in favour of the desired regioisomer 5. The unwanted regioisomer 4 may arise through either imperfect regioselectivity in the Steglich method or through acyl migration of the β-hydroxy-ester of the monoglyceride intermediate; the equilibrium position for monoglycerides of long-chain fatty acids is approximately 9:1 of the sn-1 and sn-2 regioisomers, respectively.8
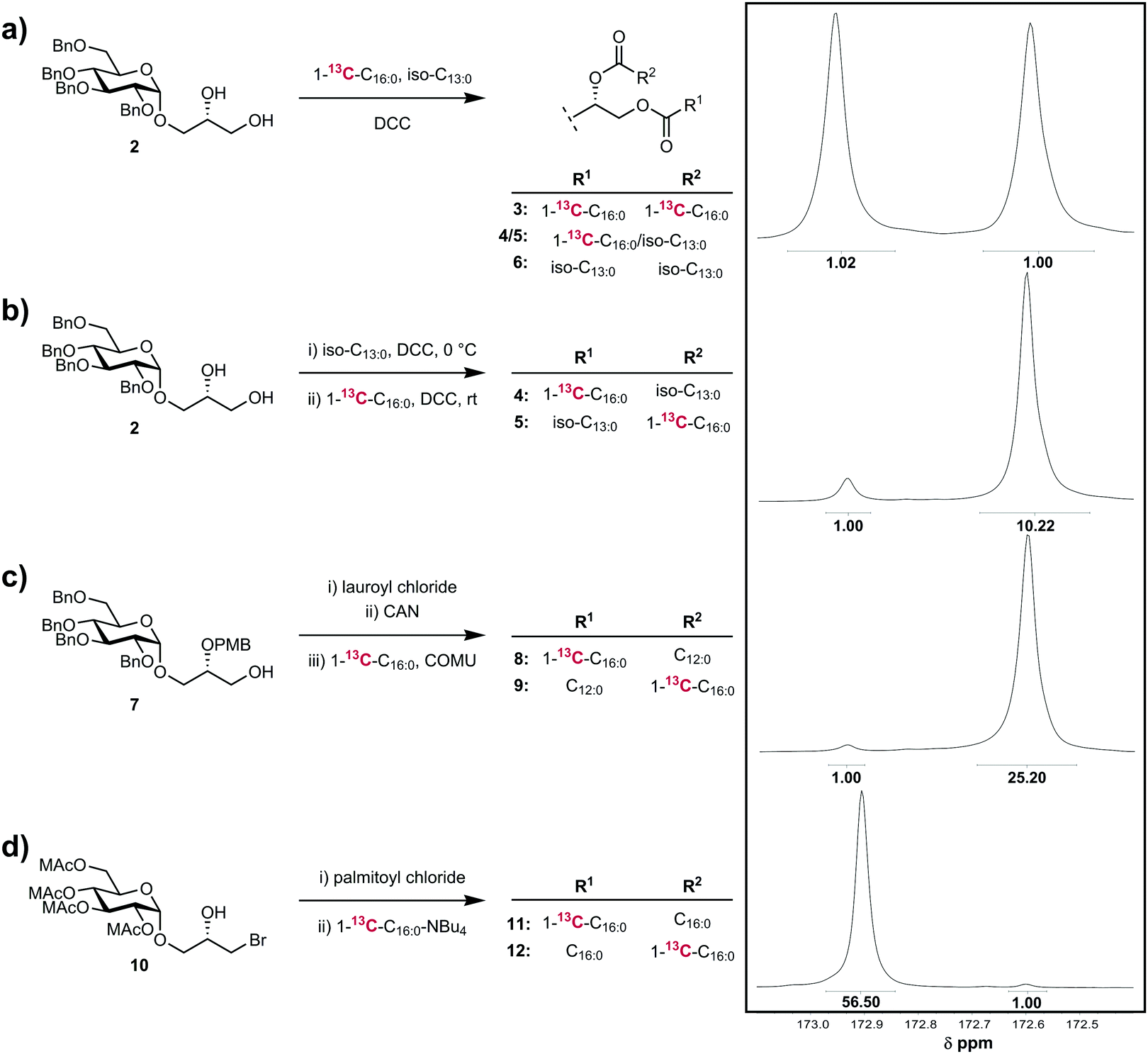 | ||
| Fig. 2 Quantitation of acylation fidelity by 13C NMR spectroscopy. (a) Acylation using a mixture of labelled and unlabelled fatty acids. (b) Stepwise acylation using Steglich esterification, with purification of the intermediate β-hydroxy-ester by flash chromatography. (c) Stepwise esterification with protecting group-control. (d) A stepwise acylation/substitution process via a β-bromoester intermediate. | ||
We next explored a stepwise acylation strategy that does not require chromatography (Fig. 2c), based on our reported approach to the α-glucuronosyl diglyceride Gl-A.11 Compound 7 was treated first with lauroyl chloride and DMAP/pyr, affording the PMB-protected monoglyceride. The PMB group was removed by treatment with CAN in CH3CN/water, and in order to limit acyl migration the crude monoglyceride, without purification, was directly acylated by treatment with 1-13C-palmitic acid and COMU/iPr2NEt in DMF. Analysis of the product revealed a ratio of sn-2 and sn-1 carbonyl resonances of 25:1, allowing calculation of R = 97.2% in favour of the desired product 9, with regioisomer 8 arising from acyl migration.
Finally, we examined a strategy that avoids β-hydroxy-ester intermediates and should provide high fidelity regioselective acylation. We previously reported an approach to prepare glycosyl diglycerides via a glycidol-derived bromohydrin 10 (MAc = methoxyacetyl) that avoids β-hydroxy ester intermediates by first esterification to afford a β-bromo-ester then bromide substitution with the carboxylate of the second fatty acid chain.16 Treatment of 10 with palmitoyl chloride and DMAP/pyr afforded an intermediate β-bromo-ester, which was purified then treated with the tetrabutylammonium salt of 1-13C-palmitic acid in toluene at reflux (Fig. 2d). Analysis of the resulting diglyceride revealed a 56:1 ratio of sn-1 and sn-2 carbonyl resonances, corresponding to R > 99%.
We applied the bromohydrin method to the total synthesis of S. pneumoniae Glc-DAG-s2 (1, Scheme 1).‡ Acylation of bromohydrin 1016 with cis-vaccenoyl chloride afforded the bromide 13. Substitution of bromide with the tetrabutylammonium salt of palmitic acid in refluxing toluene afforded 14, and deprotection of the methoxyacetyl groups using t-BuNH2 in MeOH/THF afforded Glc-DAG-s2.
 | ||
| Scheme 1 Synthesis of Streptococcus pneumonia Glc-DAG-s2. | ||
In conclusion, we report a quantitative 13C NMR method to assess the fidelity of regioselective acylation methodologies. We estimate that the method can be used to detect ∼1% of a regioisomer. We show that a stepwise Steglich procedure based on widely used literature approaches afforded diglycerides with a ratio of regioisomers of R = 92%, a composition commensurate with the known equilibrium position of monoacyl glycerols. A stepwise, protecting group controlled regioselective acylation that avoided chromatography provided better regioselective fidelity (R = 97.2%), but was surpassed by a stepwise acylation/substitution process that avoided β-hydroxy ester intermediates, and which yielded a glycosyl diglyceride that was regioselectively acylated with the limit of detection. This work should support the development of preparative methods for homogeneous diglycerides bearing two distinct acyl chains.
We thank the Australian Research Council (FT130100103, DP130102763, DP160100597) for financial support. We thank Dr Hamish Grant for technical assistance.
Notes and references
- (a) J. Zhang, C. Li, G. Yu and H. Guan, Mar. Drugs, 2014, 12, 3634 CrossRef PubMed; (b) G. Holzl and P. Dormann, Prog. Lipid Res., 2007, 46, 225 CrossRef PubMed.
- (a) D. G. Smith and S. J. Williams, Carbohydr. Res., 2016, 420, 32 CrossRef CAS PubMed; (b) J. Rossjohn, S. Gras, J. J. Miles, S. J. Turner, D. I. Godfrey and J. McCluskey, Annu. Rev. Immunol., 2015, 33, 169 CrossRef CAS PubMed.
- Y. Kinjo, P. Illarionov, J. L. Vela, B. Pei, E. Girardi, X. Li, Y. Li, M. Imamura, Y. Kaneko, A. Okawara, Y. Miyazaki, A. Gomez-Velasco, P. Rogers, S. Dahesh, S. Uchiyama, A. Khurana, K. Kawahara, H. Yesilkaya, P. W. Andrew, C. H. Wong, K. Kawakami, V. Nizet, G. S. Besra, M. Tsuji, D. M. Zajonc and M. Kronenberg, Nat. Immunol., 2011, 12, 966 CrossRef CAS PubMed.
- Y. Kinjo, E. Tupin, D. Wu, M. Fujio, R. Garcia-Navarro, M. R. Benhnia, D. M. Zajonc, G. Ben-Menachem, G. D. Ainge, G. F. Painter, A. Khurana, K. Hoebe, S. M. Behar, B. Beutler, I. A. Wilson, M. Tsuji, T. J. Sellati, C. H. Wong and M. Kronenberg, Nat. Immunol., 2006, 7, 978 CrossRef CAS PubMed.
- A. P. Uldrich, O. Patel, G. Cameron, D. G. Pellicci, E. B. Day, L. C. Sullivan, K. Kyparissoudis, L. Kjer-Nielsen, J. P. Vivian, B. Cao, A. G. Brooks, S. J. Williams, P. Illarionov, G. S. Besra, S. J. Turner, S. A. Porcelli, J. McCluskey, M. J. Smyth, J. Rossjohn and D. I. Godfrey, Nat. Immunol., 2011, 12, 616 CrossRef CAS PubMed.
- J. Wang, Y. Li, Y. Kinjo, T. T. Mac, D. Gibson, G. F. Painter, M. Kronenberg and D. M. Zajonc, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1535 CrossRef CAS PubMed.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1920, 53, 1621 CrossRef.
- F. H. Mattson and R. A. Volpenhein, J. Lipid Res., 1962, 3, 281 CAS.
- M. S. F. Lie Ken Jie, J. L. Harwood and F. D. Gunstone, in The lipid handbook, ed. F. D. Gunstone, J. L. Harwood and A. J. Dijkstra, CRC Press, Boca Raton, 2007, p. 355 Search PubMed.
- V. Pozsgay, J. Kubler-Kielb, B. Coxon, A. Marques, J. B. Robbins and R. Schneerson, Carbohydr. Res., 2011, 346, 1551 CrossRef CAS PubMed.
- B. Cao, X. Chen, Y. Yamaryo-Botte, M. B. Richardson, K. L. Martin, G. N. Khairallah, T. W. Rupasinghe, R. M. O'Flaherty, R. A. O'Hair, J. E. Ralton, P. K. Crellin, R. L. Coppel, M. J. McConville and S. J. Williams, J. Org. Chem., 2013, 78, 2175 CrossRef CAS PubMed.
- (a) G. Guella, R. Frassanito and I. Mancini, Rapid Commun. Mass Spectrom., 2003, 17, 1982 CrossRef CAS PubMed; (b) R. V. Tatituri, M. B. Brenner, J. Turk and F. F. Hsu, J. Mass Spectrom., 2012, 47, 115 CrossRef CAS PubMed; (c) J. Sauvageau, J. Ryan, K. Lagutin, I. M. Sims, B. L. Stocker and M. S. Timmer, Carbohydr. Res., 2012, 357, 151 CrossRef CAS PubMed.
- A. W. Mazur, G. D. Hiler, 2nd, S. S. Lee, M. P. Armstrong and J. D. Wendel, Chem. Phys. Lipids, 1991, 60, 189 CrossRef CAS PubMed.
- (a) J. M. Henderson, M. Petersheim, G. J. Templeman and B. J. Softly, J. Agric. Food Chem., 1994, 42, 435 CrossRef CAS; (b) P. Kalo, A. Kemppinen and I. Kilpelainen, Lipids, 1996, 31, 331 CrossRef CAS PubMed.
- M. B. Richardson, S. Torigoe, S. Yamasaki and S. J. Williams, Chem. Commun., 2015, 51, 15027 RSC.
- S. Shah, M. Nagata, S. Yamasaki and S. J. Williams, Chem. Commun., 2016, 52, 10902 RSC.
- D. J. Cookson and B. E. Smith, J. Magn. Reson., 1984, 57, 355 CAS.
- G. C. Levy and R. A. Komoroski, J. Am. Chem. Soc., 1974, 96, 678 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc09584d |
| ‡ While a previous report has disclosed an apparently synthetic version of this interesting natural product, no details for the synthetic approach were reported. See ref. 3. |
| This journal is © The Royal Society of Chemistry 2017 |