
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of axially chiral heterobiaryl alkynes via dynamic kinetic asymmetric alkynylation†
Valentín
Hornillos
*a,
Abel
Ros
ab,
Pedro
Ramírez-López
a,
Javier
Iglesias-Sigüenza
 b,
Rosario
Fernández
*b and
José M.
Lassaletta
*a
b,
Rosario
Fernández
*b and
José M.
Lassaletta
*a
aInstituto Investigaciones Químicas (CSIC-US), C/Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: vhornillos@iiq.csic.es; jmlassa@iiq.csic.es
bDepartamento de Química Orgánica, Universidad de Sevilla, C/Prof. García González, 1, 41012 Sevilla, Spain. E-mail: ffernan@us.es
First published on 11th November 2016
Abstract
The dynamic kinetic Pd0-catalyzed alkynylation of racemic heterobiaryl sulfonates was used for the asymmetric synthesis of axially chiral heterobiaryl alkynes with a broad scope. The use of Pd(OAc)2/(S)-QUINAP as the precatalyst provides products in excellent yields and enantioselectivities under mild conditions (DMSO, 40 °C). Semireduction, 1,3-dipolar cycladdition or N-oxidation served to illustrate the synthetic potential of the methodology.
Axially chiral biaryls are often found in natural products1 as well as biologically active molecules and constitute privileged frameworks for ligands in the field of asymmetric catalysis.2 Despite their key importance, methods to access these moieties in high efficiency and selectivity are still scarce and limited in substrate scope.3 While significant advances have been reported in asymmetric cross-couplings to build the central axis of the molecule,4 this direct strategy fails when heterocyclic coupling partners are employed.5 However, the growing number of applications of axially chiral 2-arylpyridines(isoquinolines) such as I–V (Fig. 1) in catalysis6 has stimulated the development of alternative strategies for their catalytic asymmetric synthesis. A handful of currently available methods include a kinetic resolution via PdII-catalyzed C–H bond iodination,7 a recently reported dynamic kinetic biocatalytic reduction of configurationally labile heterobiaryl (N-oxides) aldehydes8 and two C–C bond forming strategies: CoI or RhI-catalyzed [2+2+2] cycloadditions between nitriles and alkynes (Scheme 1a)9 and RhIII-catalyzed C–H functionalization of 1-aryl-benzo[h]isoquinolines (Scheme 1b).10 Additionally, our group reported in 2013, an alternative methodology consisting of a Pd-catalyzed dynamic kinetic asymmetric (DYKAT) Suzuki–Miyaura coupling between aryl boroxines and racemic heterobiaryl triflates (Scheme 1c),11 a strategy that was later extended to perform C–P couplings for the asymmetric synthesis of heterobiaryl phosphines including QUINAP, PINAP, and QUINAZOLINAP analogues12 and Buchwald–Hartwig aminations leading to heterobiaryl amines (Isoquinoline-Amino Naphthalene (IAN) and analogues).13
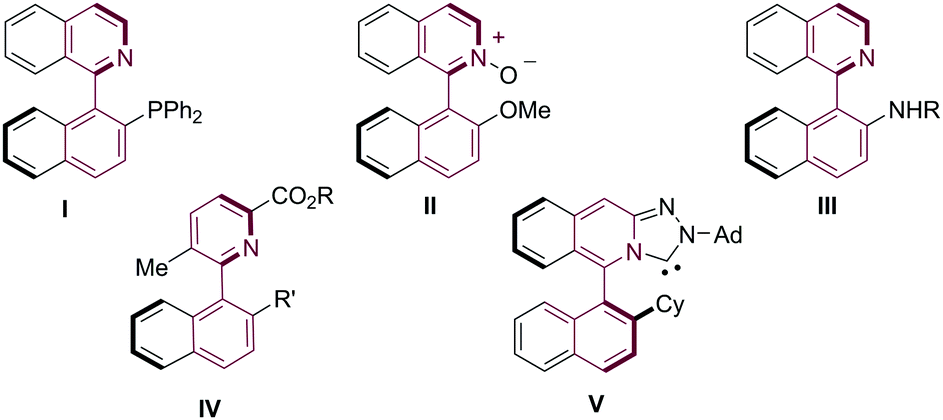 | ||
| Fig. 1 Axially chiral heterobiaryls relevant to catalysis. | ||
 | ||
| Scheme 1 C–C bond forming strategies toward axially chiral heterobiaryls. | ||
Although the dynamic kinetic Suzuki–Miyaura coupling via DYKAT represents an appealing C–C bond forming strategy for the atroposelective synthesis of axially chiral heterobiaryls, the absence of reactive or coordinating functionalities at the ortho position limits its further applicability (e.g., as chiral bidentate ligands or organocatalysts).14 We therefore examined the possibility of a DYKAT-based strategy for the introduction of versatile groups capable of further functionalization or coordination. Because alkynes are privileged building blocks,15 we decided to look into the asymmetric alkynylation of racemic heterobiaryl electrophiles (Scheme 1d).16,17
We selected the coupling of racemic 1-(isoquinolin-1-yl)naphthalene-2-yl nonaflate 1A with phenylacetylene 2a as a model reaction (Table 1). In the presence of 10 mol% of Pd(dba)2 and 11 mol% (R)-BINAP L1 and using DIPEA (3 eq.) as the base in dioxane at 60 °C, the desired product 2Aa was obtained with moderate conversion and enantioselectivity (entry 1). Although disappointing from the synthetic viewpoint, this experiment served as a proof of concept and confirmed the configurational stability of the product. A survey of different solvents revealed that DMSO facilitated high conversion while maintaining a similar enantioselectivity (entry 2). Under these conditions, different commercially available axially chiral biphosphines L2–L6 (Table 1, entries 3–7 and Table S1 in the ESI†) were examined, but the enantioselectivity could not be improved. The use of phosphoramidite ligand L7 (employed in our previous dynamic kinetic Suzuki reaction)11 and phosphino-hydrazone ligand L8 (efficient in atropo-enantioselective Suzuki–Miyaura cross-couplings)4g provided excellent conversions into the desired product 3Aa but with low enantioselectivities (entries 8 and 9). Finally we were pleased to find out that ligand L9, (S)-QUINAP L10 and the fluorinated derivative L1112a provided higher enantioselectivities with complete conversions (entries 10–12). After an additional screening of base, Pd source and temperature (entries 13–19 and Table S1 in the ESI†), we identified L10 as an optimal ligand that enables the reaction to proceed at 40 °C with a lower catalyst loading (5 mol%) and excellent results (99% conv., 97% ee). It is worth mentioning that no additional CuI co-catalyst is needed in this transformation.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L | T (°C) | Base | Solvent | Conv.b (%) | eec |
| a Conditions: 0.1 mmol of rac-1A, 0.2 mmol of alkyne. b Conversion by 1H NMR spectroscopy. c Determined by HPLC. d [Pd] (5 mol%)/L10 (6 mol%). e Pd(OAc)2 was used instead of Pd(dba)2. | ||||||
| 1 | L1 | 60 | DIPEA | Dioxane | 46 | 70 |
| 2 | L1 | 60 | DIPEA | DMSO | 90 | 68 |
| 3 | L2 | 60 | DIPEA | DMSO | 75 | 72 |
| 4 | L3 | 60 | DIPEA | DMSO | 77 | 68 |
| 5 | L4 | 60 | DIPEA | DMSO | 44 | 68 |
| 6 | L5 | 60 | DIPEA | DMSO | 90 | 18 |
| 7 | L6 | 60 | DIPEA | DMSO | 22 | 82 |
| 8 | L7 | 60 | DIPEA | DMSO | >99 | 30 |
| 9 | L8 | 60 | DIPEA | DMSO | >99 | <5 |
| 10 | L9 | 60 | DIPEA | DMSO | 73 | 90 |
| 11 | L10 | 60 | DIPEA | DMSO | >99 | 86 |
| 12 | L11 | 60 | DIPEA | DMSO | >99 | 90 |
| 13 | L9 | 50 | DIPEA | DMSO | 57 | 88 |
| 14 | L10 | 40 | DIPEA | DMSO | >99 | 94 |
| 15 | L11 | 40 | DIPEA | DMSO | >99 | 93.5 |
| 16d | L10 | 30 | DIPEA | DMSO | >99 | 93 |
| 17d | L10 | 40 | DIPEA | DMSO | >99 | 94 |
| 18d | L10 | 40 | Et3N | DMSO | >99 | 96 |
| 19d,e | L10 | 40 | Et3N | DMSO | >99 | 97 |
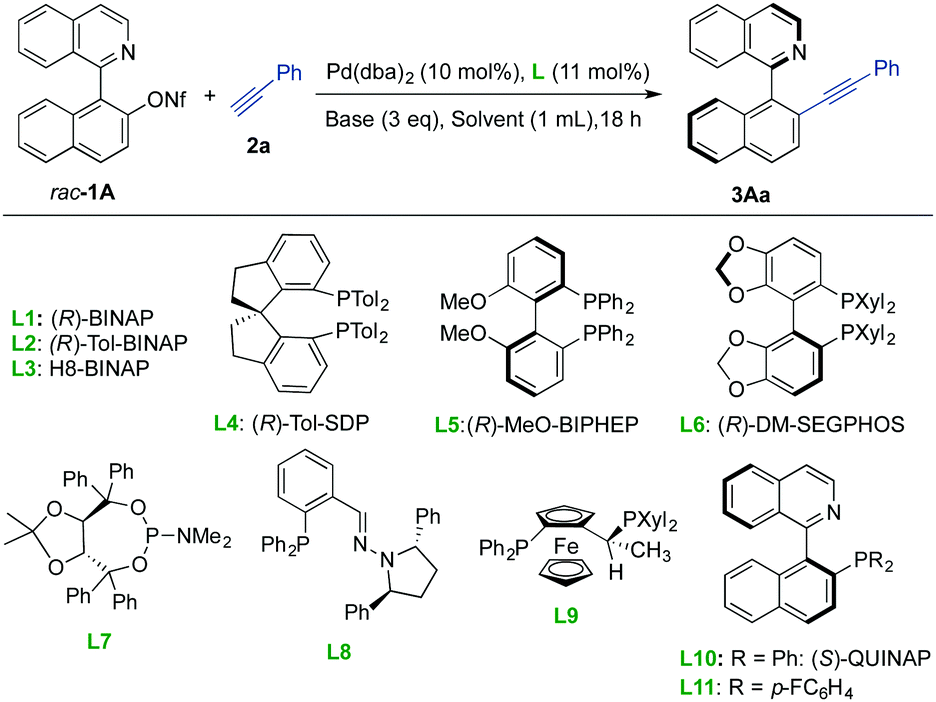
With the optimized reaction conditions in hand [5 mol% Pd(OAc)2/6 mol% QUINAP, Et3N (3 eq.) as the catalyst, DMSO, 40 °C], we next explored the alkyne scope using 1A as the heterobiaryl partner (Scheme 2). Phenylacetylenes with electron-donating (p-MeO, p-NMe2) or withdrawing substituents (p-F, p-Br) afforded the desired products (3Aa–e) in excellent yields (95% to quantitative) and enantioselectivities (up to 95% ee). Importantly, no evidence of the competing coupling at the C–Br bond was observed in the preparation of 3Ae, highlighting how the high chemoselectivity of the reaction allows for the inclusion of strategic functionalities prone to undergo further transformations. The lower enantioselectivity observed for 3Af can be attributed to the interferences that the coordination of the nitrile group, of either the starting alkyne 2f or the product, might cause in the enantiodetermining step. Excellent yields and enantioselectivities were also observed for hindered aryl- or naphthyl-substituted acetylenes 2h–k, while the use of enyne 2l and alkynes 2m–p bearing different linear and cyclic aliphatic substituents, including a secondary propargylic and homopropargylic alcohol, also provided the desired products in excellent yields and ees. The use of trimethylsilylacetylene 2q led to the TMS-protected product 3Aq, also obtained in high yield and selectivity. The ensuing high-yielding deprotection with TBAF provided the terminal alkyne 4A without erosion of optical purity. Importantly, a lower catalyst loading (1 mol% [Pd]/1.2 mol% L10) was required for the reaction of rac-1A with 2q on a larger scale (2 mmol, 1.1 g), leading to 4A in 80% overall yield and with the same ee after TMS removal.
 | ||
| Scheme 2 Scope of alkynes. All reactions were performed at 0.1 mmol scale and reached full conversion as determined by TLC and 1H NMR spectroscopy. Isolated yields after column chromatography. Ees were determined by HPLC. | ||
We next explored the scope of the reaction with respect to other heterobiaryl sulfonates using phenylacetylene 2a, 1-octyne 2n and trimethylsilylacetylene 2q as the alkyne counterparts (Scheme 3). Variations in the structure of the heterobiaryl frame did not have an impact on the reaction outcome. Thus, quinazoline and phthalazine derivatives 1B and 1C afforded the corresponding alkynes with excellent selectivities, independently of the leaving group used (OTf or ONf). As mentioned before, the corresponding TMS-protected alkynes could be desilylated using TBAF to afford the terminal alkynes 4B and 4C in high yield without compromising the configurational integrity of the final product. Additionally, heterobiaryl alkynes derived from picoline 1D could also be obtained in high yields and ees. Heterobiaryl nonaflate 1E is a priori a more challenging substrate for the undesired effects of the push–pull conjugation with the OMe group in the key oxidative addition intermediate: first, a partial double bond character and a shorter C(1)–C(1′) bond length are expected to slow down the required atropoisomerization at this stage18 (Fig. 2). Second, the dissociation of the N–Pd bond, enabling the coordination of the alkyne, is also made difficult by the higher basicity of the isoquinoline N atom. In fact, 1E was not a suitable substrate in previous DYKAT-based strategies,10,12a,13 but the better efficiency of the alkynylation reaction allowed the isolation of compounds 3Ea,n,q in excellent yields and good to excellent ees. The absolute R configurations of (R)-3Ak and (R)-3Ea were determined by X-ray diffraction analysis while those of other products 3 were assigned by analogy.
 | ||
| Scheme 3 Scope of heterobiaryls. All reactions were performed at 0.1 mmol scale and reached full conversion as determined by TLC and 1H NMR spectroscopy. Isolated yields after column chromatography. Ees were determined by HPLC. | ||
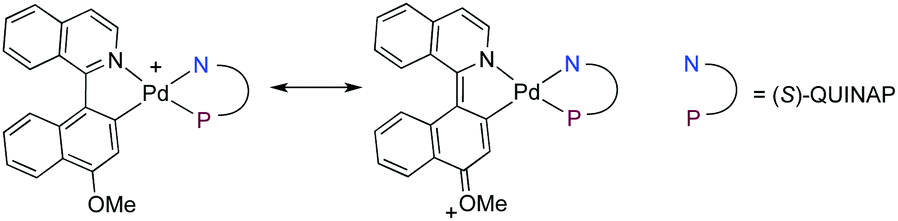 | ||
| Fig. 2 Oxidative addition intermediate from 1E. | ||
To further demonstrate the potential of this new methodology, the terminal alkyne 4A was used as a platform for the synthesis of new families of axially chiral heterobiaryls that are otherwise difficult to obtain. Highly chemoselective semireduction of the alkyne was accomplished in good yield according to a recently reported procedure19 (Scheme 4a). The resulting compound 5 represents a novel example (with axially chirality) of the so far underdeveloped chiral N/olefin hybrid ligands (e.g., OlefOx) that have already shown an excellent performance in asymmetric catalysis.20 Furthermore, a unique axially chiral N,N-ligand 6 was prepared via Cu(I)-catalyzed dipolar cycloaddition21 of 4A with benzyl azide in good yield under mild conditions (Scheme 4b). Finally, selective N-oxidation of 4A using m-CPBA afforded N-oxide 7 in 94% yield (Scheme 4c). Remarkably, the stereochemical integrity is completely preserved for all the above transformations.
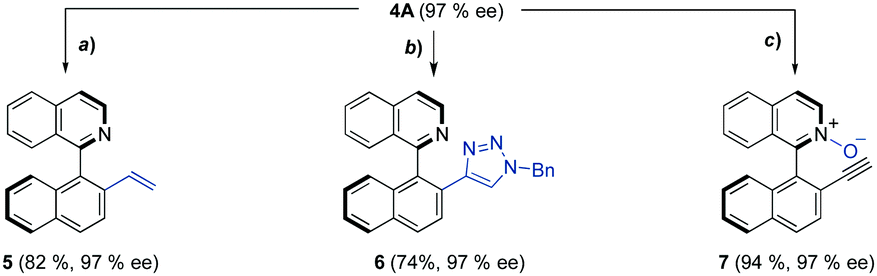 | ||
| Scheme 4 Representative derivatizations. (a) IPrCuOtBu (5 mol%), PMHS (1.2 eq.), toluene, rt, 14 h; (b) BnN3 (1.5 eq.) CuSO4 (10 mol%), sodium ascorbate (20 mol%), tBuOH/H2O, 35 °C, 5 h; (c) m-CPBA (2 eq.), THF, rt, 3 h. | ||
In summary, we have developed a highly efficient methodology for the synthesis of heterobiaryl alkynes based on a dynamic kinetic asymmetric alkynylation reaction. Broad scope, functional group tolerance and excellent enantioselectivities were achieved using a Pd0/QUINAP catalytic system (down to 1 mol%) under mild conditions (40 °C). The newly installed alkynyl group was readily transformed to access novel axially chiral bidentate ligands.
We thank the Spanish MINECO (Grants CTQ2013-48164-C2-1-P and CTQ2013-48164-C2-2-P, contract RYC-2013-12585 for A.R.), European FEDER Funds, and Junta de Andalucía (Grant 2012/FQM 10787) for financial support. VH thanks the EU 7th Framework Program, Marie Skłodowska-Curie actions for a Talent Hub fellowship (COFUND – Grant no. 291780).
Notes and references
- (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC.
- For reviews, see: (a) R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS; (b) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed; (c) H. Shimizu, I. Nagasaki and T. Saito, Tetrahedron, 2005, 61, 5405–5432 CrossRef CAS; (d) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS; (e) Y.-M. Li, F.-Y. Kwong, W.-Y. Yu and A. S. C. Chan, Coord. Chem. Rev., 2007, 251, 2119–2144 CrossRef CAS; (f) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (g) Y. Canac and R. Chauvin, Eur. J. Inorg. Chem., 2010, 2325–2335 CrossRef CAS; (h) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- (a) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) G. Ma and M. P. Sibi, Chem. – Eur. J., 2015, 21, 11644–11657 CrossRef CAS PubMed; (c) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- Seminal work and recent examples: (a) T. Hayashi, S. Niizuma, T. Kamikawa, N. Suzuki and Y. Uozumi, J. Am. Chem. Soc., 1995, 117, 9101–9102 CrossRef CAS; (b) J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 12051–12052 CrossRef CAS; (c) T. Shimada, Y.-H. Cho and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 13396–13397 CrossRef CAS PubMed; (d) K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS PubMed; (e) A. Bermejo, A. Ros, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2008, 130, 15798–15799 CrossRef CAS PubMed; (f) T. Yamamoto, Y. Akai, Y. Nagata and M. Suginome, Angew. Chem., Int. Ed., 2011, 50, 8844–8847 CrossRef CAS PubMed; (g) A. Ros, B. Estepa, A. Bermejo, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Org. Chem., 2012, 77, 4740–4750 CrossRef CAS PubMed; (h) Y. Zhou, S. Wang, W. Wu, Q. Li, Y. He, Y. Zhuang, L. Li, J. Pang, Z. Zhou and L. Qiu, Org. Lett., 2013, 15, 5508–5511 CrossRef CAS PubMed; (i) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570–573 CrossRef CAS PubMed . Recent reviews: ; (j) Y. Uozumi, Comprehensive Chirality, Elsevier, 2012, vol. 4, ch. 4.3 Search PubMed; (k) D. Zhang and Q. Wang, Coord. Chem. Rev., 2015, 286, 1–16 CrossRef CAS.
- For exceptions reaching moderate enantioselectivities see: (a) A. Mosquera, M. A. Pena, J. Pérez Sestelo and L. A. Sarandeses, Eur. J. Org. Chem., 2013, 2555–2562 CrossRef CAS; (b) K. Yamaguchi, J. Yamaguchi, A. Studer and K. Itami, Chem. Sci., 2012, 3, 2165–2169 RSC; (c) K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 RSC.
- QUINAP and analogues: (a) E. Fernández, P. J. Guiry, K. P. T. Connole and J. M. Brown, J. Org. Chem., 2014, 79, 5391–5400 CrossRef PubMed , and references cited therein; Axially chiral pyridine (isoquinoline) N-oxides: ; (b) A. V. Malkov, P. Ramírez-López, L. Biedermannová, L. Rulíšek, L. Dufková, M. Kotora, F. Zhu and P. Kočovský, J. Am. Chem. Soc., 2008, 130, 5341–5348 CrossRef CAS PubMed ; Isoquinoline-Amino Naphthalene (IAN): ; (c) S. B. Cortright and J. N. Johnston, Angew. Chem., Int. Ed., 2002, 41, 345–348 CrossRef CAS ; Axially quiral 5-methyl-(1-naphthyl)picolinic acids(esters): ; (d) S. Tanaka, T. Seki and M. Kitamura, Angew. Chem., Int. Ed., 2009, 48, 8948–8951 CrossRef CAS PubMed ; Axially chiral N-heterocyclic carbenes: ; (e) J. Francos, F. Grande-Carmona, H. Faustino, J. Iglesias-Sigüenza, E. Díez, I. Alonso, R. Fernández, J. M. Lassaletta, F. López and J. L. Mascareñas, J. Am. Chem. Soc., 2012, 134, 14322–14325 CrossRef CAS PubMed ; Axially chiral pyridoxamines: ; (f) Y. E. Liu, Z. Lu, B. Li, J. Tian, F. Liu, J. Zhao, C. Hou, Y. Li, L. Niu and B. Zhao, J. Am. Chem. Soc., 2016, 138, 10730–10733 CrossRef CAS PubMed.
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741–2745 CrossRef CAS.
- S. Staniland, R. W. Adams, J. J. W. McDouall, I. Maffucci, A. Contini, D. M. Grainger, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 10755–10759 CrossRef CAS PubMed.
- Seminal work: (a) A. Gutnov, B. Heller, C. Fischer, H. J. Drexler, A. Spannenberg, B. Sundermann and C. Sundermann, Angew. Chem., Int. Ed., 2004, 43, 3795–3797 CrossRef CAS PubMed; (b) Review: K. Tanaka, Chem. – Asian J., 2009, 4, 508–518 CrossRef CAS PubMed ; Recent examples: M. Hapke, K. Kral, C. Fischer, A. Spannenberg, A. Gutnov, D. Redkin and B. Heller, J. Org. Chem., 2010, 75, 3993–4003 Search PubMed; (c) N. Sakiyama, D. Hojo, K. Noguchi and K. Tanaka, Chem. – Eur. J., 2011, 17, 1428–1432 CrossRef CAS PubMed.
- (a) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244–13247 CrossRef CAS PubMed; (b) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed.
- A. Ros, B. Estepa, P. Ramírez-López, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2013, 135, 15730–15733 CrossRef CAS PubMed.
- (a) P. Ramírez-López, A. Ros, B. Estepa, R. Fernández, B. Fiser, E. Gómez-Bengoa and J. M. Lassaletta, ACS Catal., 2016, 6, 3955–3964 CrossRef; (b) See also: V. Bhat, S. Wang, B. M. Stoltz and S. C. Virgil, J. Am. Chem. Soc., 2013, 135, 16829–16832 CrossRef CAS PubMed.
- P. Ramírez-López, A. Ros, A. Romero-Arenas, J. Iglesias-Sigüenza, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2016, 138, 12053–12056 CrossRef PubMed.
- (a) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed; (b) Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011, ch. 4, p. 137 Search PubMed.
- (a) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (b) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed , and references cited therein.
- Reviews: (a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (b) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC; (c) J. Magano and J.-R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed; (d) M. Schilz and H. Plenio, J. Org. Chem., 2012, 77, 2798–2807 CrossRef CAS PubMed.
- For asymmetric Sonogashira couplings in other contexts see: (a) K. Kanda, T. Koike, K. Endo and T. Shibata, Chem. Commun., 2009, 1870–1872 RSC; (b) H. Zhou and Y. Uozumi, Synlett, 2013, 2550–2554 CAS.
- M. Gómez-Gallego, M. Martín-Ortiz and M. A. Sierra, Eur. J. Org. Chem., 2011, 6502–6506 CrossRef.
- (a) A. M. Whittaker and G. Lalic, Org. Lett., 2013, 15, 1112–1115 CrossRef CAS PubMed; (b) See also: K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542–1550 CrossRef CAS.
- B. T. Hahn, F. Tewes, R. Fröhlich and F. Glorius, Angew. Chem., Int. Ed., 2010, 122, 1143–1146 CrossRef PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization data. CCDC 1506065 and 1506066. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08997f |
| This journal is © The Royal Society of Chemistry 2016 |