
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution-phase synthesis of 1D tubular polymers via preorganization–polymerization†
Hao
Yu
and
Robert
Häner
*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: robert.haener@dcb.unibe.ch
First published on 21st November 2016
Abstract
The synthesis of one-dimensional tubular polymers via a preorganization–polymerization approach is presented. Prior to polymerization, photo-reactive monomers self-assemble into one-dimensional tubular structures in aqueous medium. The supramolecular polymers are subsequently converted into covalent polymers by light-induced anthracene dimerization.
The discovery of linear, one-dimensional (1D) polymers by Staudinger has greatly changed our understanding of the field.1 In conventional polymerization methods, the monomers are usually randomly oriented in solution or bulk during the polymerization process. Therefore, the growth of the polymer chains highly depends on facile access of the active chain ends to the monomers. Several factors can be detrimental to the reaction efficiency, such as uneven initiator distribution, ineffective mixing or poor heat dissipation. The polymerization reaction also tends to slow down with increasing monomer size due to steric hindrance. Moreover, undesired backbone branching caused by chain transfer can pose limitations to the physical properties of polymers. If the monomers are pre-organized in arrays with suitable orientation prior to polymerization, many of the above mentioned drawbacks can be eliminated. This preorganization–polymerization (POP) approach has been successfully applied at the air–water interface to produce two-dimensional (2D) polymers.2–4 Also, 1D and 2D polymers have been synthesized by POP in solid-state chemistry.5–10 Solution-phase POP, however, has been rarely reported11–15 and its use was often limited to cases in which the monomer preorganization relied on the addition of external agents.16–23 Supramolecular polymerization24–28 has been broadly used for the construction of sophisticated objects,29,30 including helical and tubular structures,31–33 thus providing a simple and efficient way to preorganized structures. Herein, we demonstrate a template-free, solution-phase synthesis of stable 1D tubular polymers by the POP approach. Building on the supramolecular polymerization of phosphodiester-linked aromatic chromophores reported previously,34–39 the amphiphilic 1,4-linked anthracene trimer (ANT3) is used as the building block for self-assembly of 1D tubular supramolecular polymers under aqueous conditions. The self-assembly requires no template, and the formed supramolecular tubes are subsequently converted to covalently linked polymers by UV-induced anthracene dimerization. The obtained polymers appear as aggregated fibrils of uniform height.
UV-induced chemical reactions have been widely used in synthetic chemistry.40–44 These reactions are usually fast, clean and easy to control. Therefore, photochemically reactive anthracene was chosen as the building block for the monomer.45–49 The synthesis of phosphoramidite 4, which is required for the preparation of ANT3, is summarized in Scheme 1. The starting material, 1,4-dibromoanthracene (1), was synthesized by reported procedures from 1,4-diaminoanthraquinone.50,51 The preparation of ANT3, the monomeric building block used for oligomerization, was accomplished by solid-phase synthesis. ANT3 consists of three anthracene units that are linked by alkynyl-phosphodiester linkers at positions 1 and 4. The product was purified by HPLC and characterized by mass spectrometry according to reported procedures (ESI†).
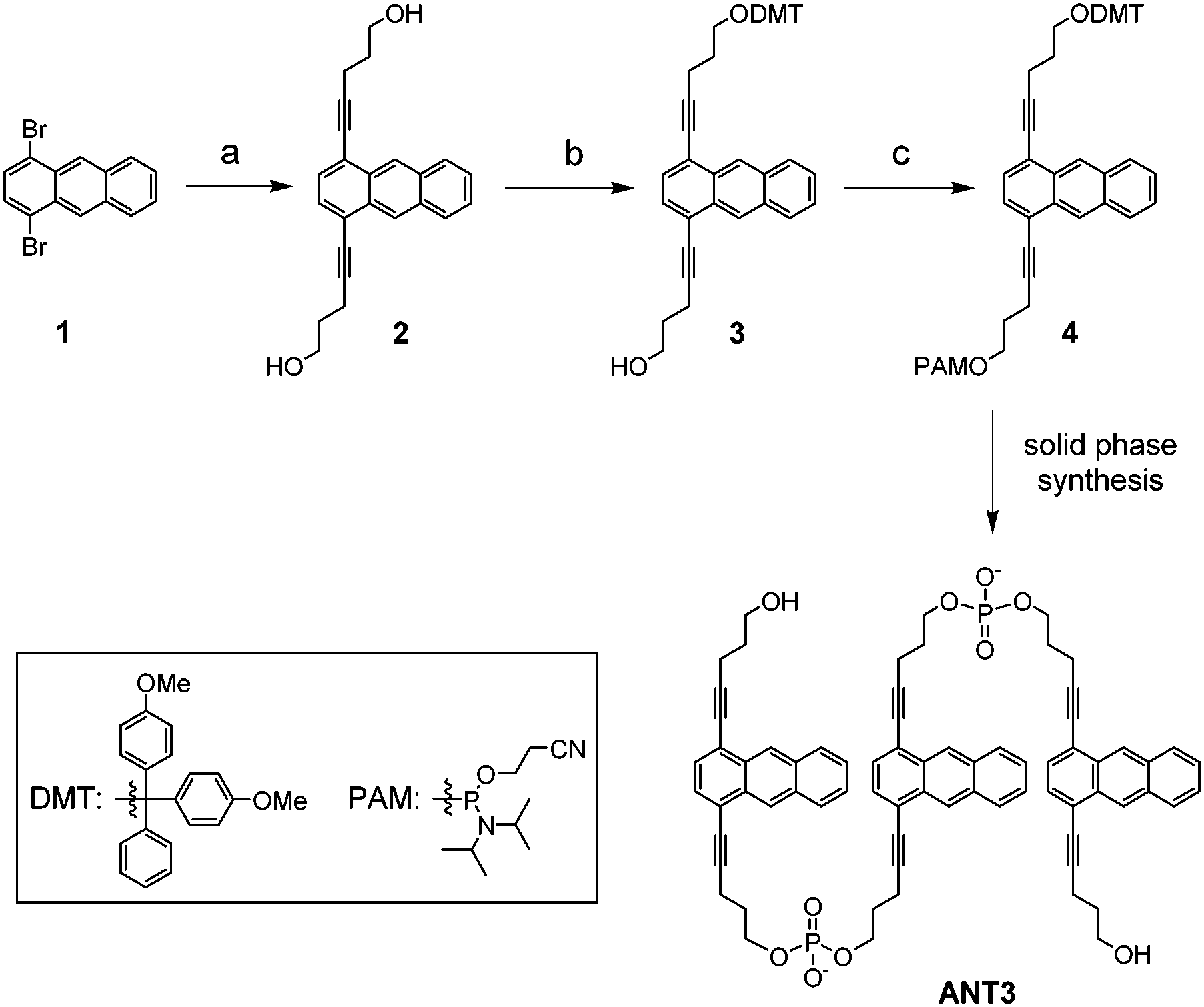 | ||
| Scheme 1 Synthetic procedure for ANT3. (a) 4-Pentyn-1-ol, CuI, Pd(PPh3)4, PPh3, triethylamine, THF, reflux, 61%; (b) DMTCl, triethylamine, DMAP, dry CH2Cl2, room temperature, 50%; (c) PAMCl, Hünig's base, dry CH2Cl2, room temperature, 74%. | ||
The supramolecular polymerization of ANT3 was conducted in an aqueous solution (10 mM NaCl and 10 mM phosphate buffer at pH 7.2) containing 5% v/v ethanol (EtOH). The mixture was heated to 80 °C and slowly cooled to room temperature over a period of 2 h. UV-vis absorption spectra of ANT3 recorded under different conditions are shown in Fig. 1a. In EtOH, the typical vibronic band pattern of anthracene was observed in the 340–430 nm range with a ratio of S0→01/S0→11 = 1.01. In aqueous solution, this ratio dropped to 0.87 and a bathochromic shift of 4 nm was observed, which indicates aromatic stacking.52,53 Furthermore, the overall intensity of the S0 → S1 transition dramatically decreased. This supports that the anthracene units are engaged in π–π stacking in aqueous solution, a prerequisite for the desired dimerization. Temperature dependent fluorescence spectra (ESI†) show that the assembly follows a nucleation/elongation mechanism.25,54
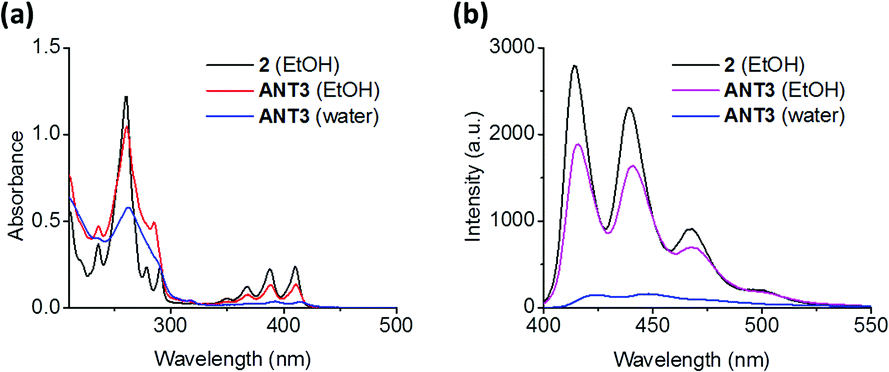 | ||
| Fig. 1 UV-vis absorption (a) and fluorescence spectra (b) of 2 and ANT3 in EtOH, and ANT3 in aqueous buffer (10 mM sodium chloride, 10 mM sodium phosphate at pH = 7.2, with 5% v/v EtOH); concentrations: 2 (3 μM), ANT3 (1 μM). | ||
The emission spectrum (excitation at 390 nm) of ANT3 in aqueous solution does not show the presence of an anthracene excimer (Fig. 1b). Compared with the fluorescence spectrum of ANT3 in EtOH, a large decrease of the intensity is observed, which is further evidence for the formation of anthracene aggregates in water.
Photodimerization of anthracene was carried out by irradiation at 366 nm under an argon atmosphere (ESI†). The anthracene absorption decreased over time and no further changes were observed after 150 min (Fig. 2a). Based on the absorption change at 395 nm, the monomer consumption was approx. 70%. The incompleteness of the reaction can be rationalized by the following model. After ANT3 units are positioned within the supramolecular polymers (Fig. 2b), two extreme scenarios for the photopolymerization exist, assuming that there are no non-linked neighbouring anthracenes (or defects) left after irradiation. Path 1 (100% dimerization) would obviously leave no unreacted anthracenes. Path 2, on the other hand, illustrates the minimal degree of photodimerization: every third consecutive anthracene remains unreacted, thus leaving the maximum number of isolated anthracenes incapable of further reaction. This scenario leads to a 67% dimerization degree, which, although not complete, results in a complete linkage of neighbouring ANT3 units in the supramolecular chain. Thus, any anthracene consumption equal to or higher than 67% indicates a complete conversion of a supramolecular chain into a covalent polymer. The experimentally observed value of 70% lies close to the theoretical value of path 2, suggesting that approximately one-third of the anthracenes remains unreacted during the dimerization reaction.
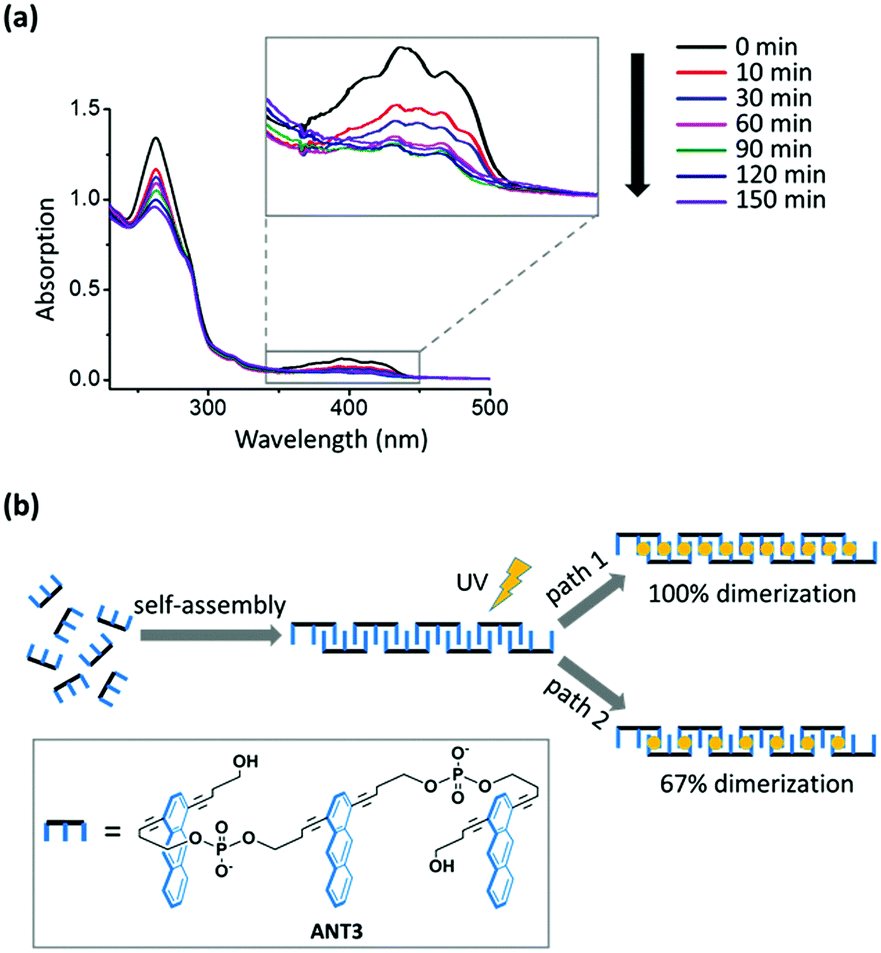 | ||
| Fig. 2 (a) Monitoring of anthracene dimerization within supramolecular polymers by changes in the UV-vis absorption. Inset: Anthracene absorption continuously decreases over time until a minimum value is reached after 150 min. For conditions, see Fig. 1. (b) Illustration of two extreme reaction pathways leading to covalent linear polymers. Assuming that defects (i.e. non-linked anthracene neighbors) are absent, paths 1 and 2 represent the theoretical maximum (100%) and the required minimum (67%) of the dimerization degree in the polymer chain. | ||
The samples before and after UV irradiation were examined by tapping-mode AFM experiments after deposition on 3-aminopropyltriethoxy silane-modified mica. Typical images are shown in Fig. 3. Before UV irradiation, ANT3 forms 1D supramolecular polymers (1D-SP) in aqueous solution. The SPs appear as aggregated fibrils with a uniform height of ∼4 nm (Fig. 3a). In 90% EtOH, ANT3 is completely dissolved and no aggregates are observed. No significant changes in the appearance of the polymers are observed after conversion of the 1D-SPs into 1D polymers (1DP) by covalent linking in water (Fig. 3c). This is not surprising because the aggregates are expected to retain their overall shape regardless of the formation of covalent bonds. However, transferring the polymers into 90% ethanol leads to substantial morphological changes. As shown in Fig. 3d, the polymers form extended aggregates, in which single 1DPs in the micrometer range are clearly visible. This confirms a highly efficient conversion of 1D-SP into 1DPvia anthracene dimerization because non-covalently linked units would disassemble in ethanol leading to fragmentation of the chains.
 | ||
| Fig. 3 AFM images of ANT3 polymers. Before irradiation: (a) 1D-SPs formed by ANT3 self-assembly in water. (b) No aggregates are formed in EtOH. After irradiation: (c) 1DP in water. (d) Aggregated 1DP in EtOH. | ||
Based on the proposed interdigitated model (see above, Fig. 2b), the polymers should have a nominal height of ∼2 nm, corresponding to the dimensions of ANT3 as illustrated in Fig. 4. The experimentally determined height of 4 nm accounts for a double layer, which indicates that the polymers exist as nanotubes in solution and, after deposition on mica, are observed as collapsed, double layered objects. Even though we cannot rule out alternative assembly pathways, the tubular structures are best rationalized31,32 by a helical arrangement of the interdigitated ANT3 polymers. The wall of the helical tube possesses a sandwich-type structure. The middle hydrophobic anthracene layer is well shielded from the solvent by the two surrounding hydrophilic layers.
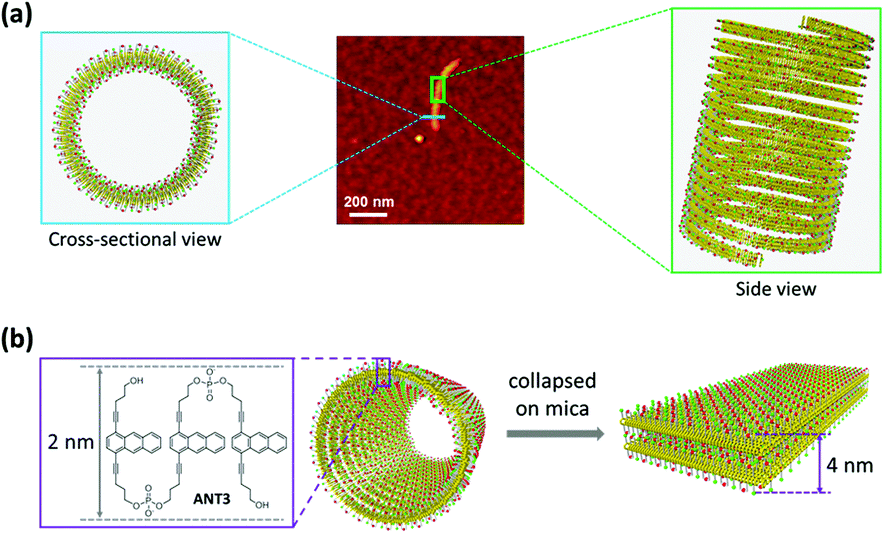 | ||
| Fig. 4 (a) Illustration of a tubular structure formed by helical arrangement of polymerized ANT3. Middle: AFM image of a single polymer. Left and right: Cross-section and side view of the tube. (b) Adsorption on mica leads to the collapse of the tube and formation of a double layered structure (4 nm). | ||
The tubular shape of both supramolecular and covalent polymers is further confirmed by TEM experiments (Fig. 5 and ESI†). The width of the flattened tubular 1D-SP was measured to be 12–13 nm. According to the proposed helical structure, this translates into approximately 25 ANT3 units constituting one turn of the helix. Assuming the pitch is 1 nm, polymers with a length of 1 μm consist of 2.5 × 104ANT3 units (see the ESI† for detailed calculations).
 | ||
| Fig. 5 (a) TEM image of 1D-SP obtained from aqueous solution. (b) Selected area from (a) recorded with higher resolution (width of the tube = 12–13 nm). (c) and (d): TEM images of 1DP obtained from aqueous solution. Conditions: 10 mM sodium chloride, 10 mM sodium phosphate at pH = 7.2, with 5% EtOH; ANT3 concentrations: 0.1 μM. | ||
In conclusion, the synthesis of a linear polymer using a novel solution-phase POP approach has been described. AFM and TEM experiments unambiguously confirmed the formation of one-dimensional supramolecular polymers, as well as their subsequent internal linking via covalent bond formation. The polymers appear as aggregated fibrils with a length of up to several micrometers. We propose a tubular structure for both supramolecular and covalent polymers. The combination of monomer preorganization and light-induced covalent linkage avoids common drawbacks (e.g. requirement for an initiator or a catalyst; branch formation) inherently associated with conventional bulk or solution-phase polymerization. This method may serve as a general way to prepare other types of polymers with different structures and functionalities.
This work was supported by the Swiss National Foundation (Grant 200020_149148). Transmission Electron Microscopy was performed on equipment supported by the Microscopy Imaging Center (MIC), University of Bern, Switzerland.
Notes and references
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53B, 1073–1085 CrossRef CAS
.
- D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450–3453 CrossRef CAS PubMed
- P. Payamyar, K. Kaja, C. Ruiz-Vargas, A. Stemmer, D. J. Murray, C. J. Johnson, B. T. King, F. Schiffmann, J. VandeVondele, A. Renn, S. Götzinger, P. Ceroni, A. Schütz, L. T. Lee, Z. Zheng, J. Sakamoto and A. D. Schlüter, Adv. Mater., 2014, 26, 2052–2058 CrossRef CAS PubMed
- W. Y. Dai, F. Shao, J. Szczerbinski, R. McCaffrey, R. Zenobi, Y. H. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed
- D. Y. Sun and C. A. Reed, Chem. Commun., 2000, 2391–2392 RSC
- P. Kissel, R. Erni, W. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Goetzinger, A. D. Schlüter and J. Sakamoto, Nat. Chem., 2012, 4, 287–291 CrossRef CAS PubMed
- M. J. Kory, M. Woerle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed
- X. Ouyang, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2003, 125, 12400–12401 CrossRef CAS PubMed
- A. Matsumoto, T. Odani, K. Sada, M. Miyata and K. Tashiro, Nature, 2000, 405, 328–330 CrossRef CAS PubMed
- H. Ihara, M. Takafuji, C. Hirayama and D. F. O'Brien, Langmuir, 1992, 8, 1548–1553 CrossRef CAS
- S. B. Lee, R. Koepsel, D. B. Stolz, H. E. Warriner and A. J. Russell, J. Am. Chem. Soc., 2004, 126, 13400–13405 CrossRef CAS PubMed
- W. Jin, T. Fukushima, A. Kosaka, M. Niki, N. Ishii and T. Aida, J. Am. Chem. Soc., 2005, 127, 8284–8285 CrossRef CAS PubMed
- J. Motoyanagi, T. Fukushima, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 4220–4221 CrossRef CAS PubMed
- T. Yamamoto, T. Fukushima, Y. Yamamoto, A. Kosaka, W. Jin, N. Ishii and T. Aida, J. Am. Chem. Soc., 2006, 128, 14337–14340 CrossRef CAS PubMed
- K. Sada, M. Takeuchi, N. Fujita, M. Numata and S. Shinkai, Chem. Soc. Rev., 2007, 36, 415–435 RSC
- J. F. Xu, Y. Z. Chen, L. Z. Wu, C. H. Tung and Q. Z. Yang, Org. Lett., 2013, 15, 6148–6151 CrossRef CAS PubMed
- Q. Zhang, D. H. Qu, J. Wu, X. Ma, Q. Wang and H. Tian, Langmuir, 2013, 29, 5345–5350 CrossRef CAS PubMed
- P. Wei, X. Yan and F. Huang, Chem. Commun., 2014, 50, 14105–14108 RSC
- X. Zhang, Y. Gao, Y. Lin, J. Hu and Y. Ju, Polym. Chem., 2015, 6, 4162–4166 RSC
- T. Hatano, M. Takeuchi, A. Ikeda and S. Shinkai, Org. Lett., 2003, 5, 1395–1398 CrossRef CAS PubMed
- T. Hatano, A. H. Bae, M. Takeuchi, A. Ikeda and S. Shinkai, Bull. Chem. Soc. Jpn., 2004, 77, 1951–1957 CrossRef CAS
- T. Hatano, A. H. Bae, M. Takeuchi, N. Fujita, K. Kaneko, H. Ihara, M. Takafuji and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 465–469 CrossRef CAS PubMed
- T. Aida, E. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed
- Z. Xie, H. He, Y. Deng, X. Wang and C. Liu, J. Mater. Chem. C, 2013, 1, 1791–1797 RSC
- V. Croué, S. Goeb, G. Szaloki, M. Allain and M. Sallé, Angew. Chem., Int. Ed., 2016, 55, 1746–1750 CrossRef PubMed
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS PubMed
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS PubMed
- H. J. Kim, W. C. Zin and M. Lee, J. Am. Chem. Soc., 2004, 126, 7009–7014 CrossRef CAS PubMed
- A. V. Rudnev, V. L. Malinovskii, A. L. Nussbaumer, A. Mishchenko, R. Häner and T. Wandlowski, Macromolecules, 2012, 45, 5986–5992 CrossRef CAS
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2014, 53, 13609–13613 CrossRef CAS PubMed
- M. Vybornyi, Y. B.-C. Hechevarria, M. Glauser, A. V. Rudnev and R. Häner, Chem. Commun., 2015, 51, 16191–16193 RSC
- Y. Vyborna, M. Vybornyi, A. V. Rudnev and R. Häner, Angew. Chem., Int. Ed., 2015, 54, 7934–7938 CrossRef CAS PubMed
- Y. Vyborna, M. Vybornyi and R. Häner, J. Am. Chem. Soc., 2015, 137, 14051–14054 CrossRef CAS PubMed
- C. D. Bösch, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2016, 55, 9961–9964 CrossRef PubMed
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010 Search PubMed
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC
- R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed
- M. O'Donnell, Nature, 1968, 218, 460–461 CrossRef
- E. J. Bowen and D. W. Tanner, Trans. Faraday Soc., 1955, 51, 475–481 RSC
- F. D. Greene, S. L. Misrock and J. R. Wolfe, J. Am. Chem. Soc., 1955, 77, 3852–3855 CrossRef CAS
- H. Bouas-Laurent, A. Castellan, J. P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC
- S. E. Wheeler, A. J. McNeil, P. Müller, T. M. Swager and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 3304–3311 CrossRef CAS PubMed
- B. VanVeller, D. Robinson and T. M. Swager, Angew. Chem., Int. Ed., 2012, 51, 1182–1186 CrossRef CAS PubMed
- A. E. Clark, C. Y. Qin and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 7586–7595 CrossRef CAS PubMed
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed
- A. L. Nussbaumer, F. Samain, V. L. Malinovskii and R. Häner, Org. Biomol. Chem., 2012, 10, 4891–4898 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and analytical details; additional spectra and images. See DOI: 10.1039/c6cc08650k |
| This journal is © The Royal Society of Chemistry 2016 |