
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical CO2 reduction in water using a co-immobilised nickel catalyst and a visible light sensitiser†
Gaia
Neri
ab,
Mark
Forster
ab,
James J.
Walsh
ab,
C. M.
Robertson
a,
T. J.
Whittles
bc,
Pau
Farràs
 d and
Alexander J.
Cowan
*ab
d and
Alexander J.
Cowan
*ab
aDepartment of Chemistry, University of Liverpool, L69 7ZD, UK. E-mail: acowan@liv.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, L69 7ZD, UK
cDepartment of Physics, University of Liverpool, L69 7ZE, Liverpool, UK
dDepartment of Chemistry, National University of Ireland Galway, Galway, Ireland
First published on 15th November 2016
Abstract
A dye-sensitised CO2 reduction photocatalyst that operates in water is reported. Transient spectroscopy demonstrates that the facile co-immobilisation of a Ru dye and a Ni CO2 reduction electrocatalyst enables efficient on-particle electron transfer leading to photocatalytic activity that greatly exceeds the equivalent solution based system.
The successful development of materials that can efficiently and cost-effectively enable solar fuel production via the reduction of CO2 and oxidation of water would have a major impact upon the energy landscape. Despite progress in recent years relatively few CO2 reduction photocatalysts that operate efficiently in water are known.1–4 Instead many CO2 reduction photocatalysts are studied in non-aqueous solvents as the increased solubility of CO2 and decreased proton concentration minimises hydrogen evolution, a competitive reduction reaction which occurs at a similar potential to CO2 reduction (e.g. CO2 + 2e− + 2H+ → CO + H2O, E0SHE = −0.12 V).1
A promising approach to achieving CO2 reduction in water is to couple a molecular electrocatalyst to a light absorbing semiconductor or dye molecule that is able to generate sufficiently reducing photoelectrons for transfer to the catalytic centre.5 This approach has been demonstrated by several groups with a number of studies exploring the coupling of Ru and Re diimine catalysts to a series of different light absorbers including TaON, InP and g-C3N4 to achieve visible light driven CO2 reduction in water.2,6,7 Ni and Co cyclams (cyclam = 1,4,8,11-tetraazacyclotetradecane) stand out amongst the known electrocatalysts for CO2 reduction in water as they are highly selective, have relatively low onset potentials and are based only on abundant elements.8,9 However, to date, attempts to use NiCyc (NiCyc = Ni(cyclam)2+) in a photochemical system have demonstrated mixed results. Some studies examined the use of p-type photoelectrodes such as Si, GaP or GaAs with NiCyc in solution; however activity was typically short-lived and a significant (<−0.6 VNHE) applied bias was still required.10,11 An alternative photocatalytic approach uses [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in aqueous solution with NiCyc.12–15 Here excitation of the ruthenium dye with visible light gives rise to a metal to ligand charge transfer (MLCT) state16 that is reductively quenched by a sacrificial electron donor, such as ascorbate, to form [Ru(bpy)3]+ that in turn transfers an electron to NiCyc (Scheme 1a).13 However initial studies reported low efficiencies, proposed to be due to inefficient electron transfer from the reduced ruthenium dye to NiCyc. Several groups have since developed elegant supramolecular sensitiser/catalyst assemblies17–19 (Scheme 1b) with the aim of facilitating electron transfer, although to the best of our knowledge none of these systems have shown an increase in activity. This approach also requires the synthesis of complex ligands and linkers which must be redesigned for each catalyst-sensitiser combination. Co-localization of a chromophore and catalytic centre on a photochemically inert support has been shown to be a facile route to active water splitting materials.20,21 We hypothesised that such an approach would also be applicable to achieving efficient CO2 reduction using an immobilised dye and a NiCyc derivative (Scheme 1c), with the close proximity of components enabling efficient on-particle electron transfer.
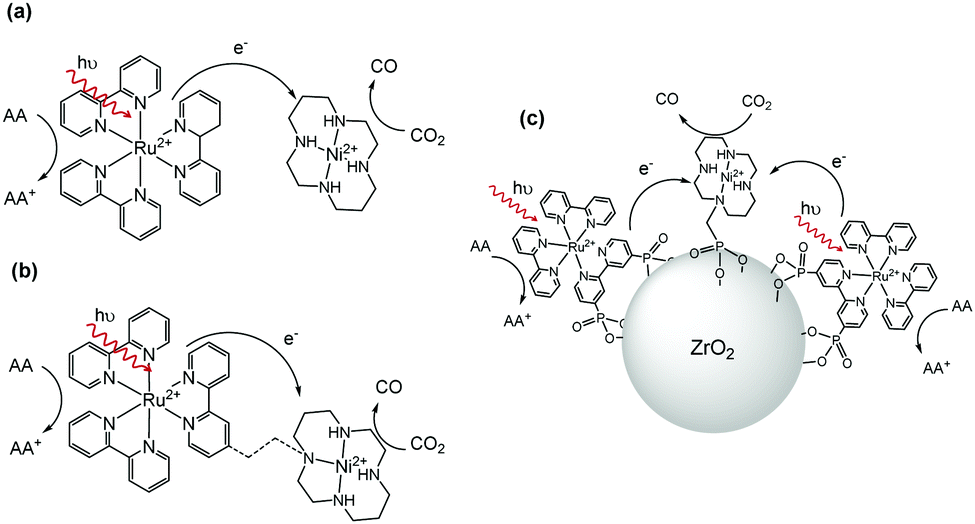 | ||
| Scheme 1 [Ru(bpy)3]2+ sensitised CO2 reduction using NiCyc in solution (a) is limited in part by low electron transfer rates. A simple alternative to synthetically complex supramolecular systems (b) is to co-immobilise the catalytic components on a photochemically inert support (c). The dashed line in (b) represents an undefined covalent linkage. | ||
We previously reported22 the surface immobilisation of a carboxylic acid functionalised NiCyc on TiO2; however the catalyst desorbed in water and CO2. Phosphonate linkages to metal oxide surfaces are known to be more stable in aqueous environments.23 Therefore CycP (CycP = [(1,4,8,11-tetraazacyclotetradecan-1-yl)methylene]phosphonic acid) was synthesised according to literature methods24 and complexed to NiCl2 to yield the novel catalyst NiIICycP (synthetic details in ESI,† structure in Fig. 1 inset). Electrochemical studies of NiCycP in solution confirm that the addition of the pendant phosphonate group does not prevent electrocatalytic CO2 reduction, Fig. 1. Using a hanging mercury drop electrode (HMDE) in 0.1 M NaClO4 (pH 4) with 1 mM NiCycP under argon an irreversible NiII/I reduction at −1.23 VNHE, similar to the NiII/I potential of the unsubstituted NiCyc (−1.30 VNHE)8 is observed, Fig. 1a. At potentials negative of −0.95 VNHE a large increase in current for NiCycP occurs under CO2 due to electrocatalytic CO2 reduction. The ratio of the peak current in the presence (icat) and absence (ip) of CO2 (icat/ip) is a commonly employed measure of catalytic activity and for NiCycP icat/ip = 19, compared to NiCyc icat/ip = 31.22
 | ||
Fig. 1 (a) Cyclic voltammetry of NiCycP in 0.1 M NaClO4 (pH 4) at 100 mV s−1 on a HMDE under Ar and CO2. The structure of NiCycP is shown in the inset. (b) Linear sweep voltammetry of ZrO2 (dashed lines) and ZrO2/NiCycP (solid lines) films in CH3CN/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) 0.1 M TBAPF6 at 100 mV s−1 under CO2 and Ar. :1) 0.1 M TBAPF6 at 100 mV s−1 under CO2 and Ar. | ||
NiCycP was immobilised onto ZrO2 nanoparticles (∅ ∼ 100–20 nm) by soaking in an ethanolic solution of NiCycP for 48 hours, prior to washing with ethanol to remove unbound NiCycP (details in ESI† 1.2). FTIR and XPS spectroscopies confirm the successful formation of ZrO2/NiCycP (Fig. S5–S10, ESI†) with binding occurring through the phosphonate linkage. ICP measurements demonstrated that soaking of samples in aqueous solution (pH 4) led to no loss of catalyst into solution confirming the stability of the phosphonate linkage (Table S1, ESI†). Electrochemical studies of ZrO2/NiCycP on FTO glass were also carried out to confirm that the immobilised catalyst remains electrochemically active, Fig. 1b. In argon-purged CH3CN with 10% water a cathodic current at potentials negative of −0.8 VNHE (Fig. 1(b)) is observed from an unmodified ZrO2 electrode in-line with previous studies which have shown the availability of electroactive states at potentials up to 0.5 V below the conduction band edge (ca. −1.2 VNHE at pH 4).25 With a ZrO2/NiCycP electrode a reduction feature is observed at ca. −0.9 VNHE which is assigned to the NiII/I couple. Under CO2 the reduction shifts anodically to ca. −0.8 VNHE. From these measurements we estimate a driving force for electron transfer from the known phosphonated ruthenium bipyridine dye, RuP (RuP = [RuII(2,2’-bipyridine)2(2,2’-bipyridine-4,4’-diylbis(phosphonic acid))]) to immobilised NiCycP to be ca. −0.3 eV under CO2 (RuP/RuP− = −1.1 VNHE).
ZrO2/RuP/NiCycP photocatalysts were prepared by soaking ZrO2 nanoparticles in ethanolic solutions of RuP and NiCycP for 48 hours (full details in ESI† 1.2). The presence of both NiCycP and RuP on ZrO2 was confirmed through FTIR, UV-Vis, ICP and X-ray photoelectron spectroscopies (Fig. S2–S10, ESI†) and quantified by ICP analysis (Table S1, ESI†). Photocatalytic experiments were carried out using 2 mg of ZrO2/RuP/NiCycP in 2 ml of CO2-purged ascorbate buffer (pH 4) under illumination (λ = 375–795 nm, 40 mW cm−2 incident). As the light-driven reduction of CO2 to CO is a multi-step process requiring the delivery of two electrons to NiCycP per catalytic cycle, it may be advantageous to have multiple light absorbers per catalytic centre.26 Therefore we initially screened a number of RuP:NiCycP ratios for CO2 reduction (Fig. S11, ESI†). This led to the identification of an optimum on particle RuP:NiCycP ratio of ca. 2.6:1, achieved by the use of a 5:1 ([RuP]:[NiCycP], 0.5 mM:0.1 mM) ethanolic solution. The ability to rapidly screen multiple catalyst and sensitiser ratios highlights an important advantage of the simple on-particle assembly approach.27 This initial communication focuses on the mechanistic validation of the design approach to CO2 reduction, however future work will expand this screening study to also explore the optimization of light driven NiCycP systems using scalable visible light sensitisers. For the ZrO2/RuP/NiCycP (2.6:1) photocatalyst we measured photocatalytic CO and H2 evolution rates of 2.0 (±0.2) μmol g−1 h−1 and 8.3 (±1.3) μmol g−1 h−1 respectively, normalised for the total mass of the photocatalyst. Considering that the ZrO2 is an inert support a more appropriate measure of activity may be to only consider the mass of the photo/catalytically active materials (RuP and NiCycP), giving rise to a relatively high CO evolution rate of 322 (±26) μmol h−1 g−1(RuP, NicycP) (incident light intensity 40 mW cm−2), Fig. 2. Within 7 hours a turnover number per NiCycP (TONNi) of 4.8 for CO production was achieved. At prolonged periods activity decreased, presumably due to the reported deactivation pathway of NiICyc-CO formation;28 however the TON in the on-particle system still greatly exceeds that previously reported for a solution based approach using NiCyc (TONNi ∼ 0.1).13 The achieved selectivity H2:CO (4.15:1) is relatively low, but it does exceed that originally reported for NiCyc in solution at pH 4 (6.4:1).13 To confirm that visible light driven CO2 reduction is occurring a number of control experiments were carried out, Fig. 2 and Fig. S12–S14 (ESI†). Firstly experiments in the absence of CO2 (under argon) yielded no significant quantity of CO on the timescales studied, Fig. S13 (ESI†). Isotopic labelling measurements using 13CO2 also demonstrated the formation of 13CO, Fig. S12 (ESI†). Experiments in the absence of either the RuP or NiCycP also led to a large loss in activity for CO production, in-line with the proposed mechanism in Scheme 1c being the dominant pathway, Fig. 2. Finally experiments using a 420 long-pass filter showed that CO production is maintained, Fig. S14 (ESI†). Strikingly, control experiments using equivalent quantities of NiCycP (6.8 nmol, 3.4 μM) and RuP (17.8 nmol, 8.9 μM) in a 2 ml 0.1 M ascorbate solution (pH 4, CO2 purged) showed a ×30 decrease in activity compared to the equivalent ZrO2/RuP/NiCycP sample (Fig. 2) with a TONNi = 0.16 for CO production after 7 hours. It is therefore apparent that the activity of the on-particle system greatly exceeds the equivalent solution based system.
 | ||
| Fig. 2 Rate of photocatalytic CO2 reduction to CO normalised for the combined mass of RuP and NiCycP in each experiment. In a typical experiment 2 mg of ZrO2/RuP/NiCycP in 2 ml of CO2 saturated 0.1 M ascorbate (pH 4) is irradiated with 375–795 nm light, 40 mW cm−2 for 7 hours. In experiment 5 a solution of NiCycP (3.4 μM) and RuP (8.9 μM) (0.1 M ascorbate (pH 4)), containing an equivalent Ni and Ru content to experiment 2, was used. | ||
For the remainder of the communication we have used transient absorption (TA) and time resolved photoluminescence (TR-PL) spectroscopy to explore the hypothesis that the enhanced photocatalytic activity for the on-particle is due to improved electron transfer from photogenerated RuP− to NiCycP, Scheme 2. Steady state emission spectroscopy of RuP/ZrO2 films excited at 435 nm showed the formation of the MLCT excited state (RuP*, λem = 618 nm)16 which was partially quenched in the presence of 0.1 M ascorbate (pH 4), Fig. S18 (ESI†). In line with past reports we measured a quenching lifetime of ca. 310 ns by TR-PL, Fig. S17 (ESI†).21 TA spectroscopy of ZrO2/RuP in water (pH 4) following 355 nm excitation showed the formation of a ground state bleach (400–500 nm) and an excited state absorption feature ca. 625 nm that was heavily overlapped with the large negative feature from 550 nm to 750 nm due to the strong emission of RuP* (Fig. S15a, ESI†).23 In-line with past reports we see no evidence for the formation of RuP+ (λmax = 700 nm) confirming that electron injection into ZrO2 cannot occur from the dye excited state (RuP+/RuP* = −0.95 VNHE).29 TA spectra of ZrO2/RuP (Fig. S15b, ESI†) and ZrO2/RuP/NiCycP (Fig. 3a) in 0.1 M ascorbate solution both showed similar features with a bleach at 425 nm, corresponding to the loss of the ground state, and a positive absorption at ca. 510 nm that can be readily assigned to RuP−21 overlapped with a broad negative feature (λmaxca. 650 nm) due to the remaining emission from the MLCT excited state. TA spectroscopy therefore confirms that reductive quenching of a fraction of the RuP* population by ascorbate is occurring. The decay of the 510 nm feature of ZrO2/RuP− is well fitted to a single stretched exponential function of the form ΔOD510nm = y0 + A1e(−kt)β with an apparent first order rate constant kapp = 1.4 × 103 s−1 (Fig. 4a, see ESI† Section 3 for the full kinetic model). It has been shown that electron injection21 into the ZrO2 conduction band does not occur from RuP− therefore it is proposed that the loss of RuP− in ZrO2/RuP is primarily due to the back reaction with oxidised ascorbate species.17 Significantly we find that the rate of RuP− decay is greatly accelerated with ZrO2/RuP/NiCycP (kapp = 7.8 × 103 s−1). Under CO2 no change in the rate of electron transfer to NiCycP was observed (Fig. 3b). The weak extinction coefficient of NiCyc and its related complexes (e.g. NiCycP in solution (aq), λmax = 344 nm (24 M−1cm−1) and 533 nm (12 M−1cm−1)), coupled to the spectral range of our TA instrument (λprobe 450–950 nm), prevents direct observation of the formation of NiCycP− (ca. 390 nm).17 Nonetheless the accelerated rate of decay of RuP− upon co-immobilisation of NiCycP strongly indicates that efficient electron transfer can occur from RuP− to NiCycP, in line with the calculated driving force for this process (ca. −0.3 eV).
 | ||
| Scheme 2 Kinetic scheme for the light driven reduction of NiCycP leading to CO2 reduction using on particle ZrO2/RuP/NiCycP compared to the dye and catalyst in solution. Lifetimes of exponential fits from TA and emission studies are given in bold. ‡ Values taken from ref. 21. | ||
 | ||
| Fig. 3 TA spectra of ZrO2/RuP/NiCycP in the presence of 0.1 M ascorbate (pH = 4) under argon and under CO2, following 355 nm (6 ns) excitation at the time delays indicated. Both spectra show the presence of a transient feature centred at ca. 510 nm assigned to RuP−. | ||
 | ||
| Fig. 4 (a) TA kinetic traces of RuP− (510 nm) showing the accelerated decay of RuP− for ZrO2/RuP/NiCycP in 0.1 M ascorbate following 355 nm (6 ns) excitation. (b) Kinetic trace of RuP− in solution following the 355 nm excitation of RuP (8.9 μM) in 0.1 M ascorbate in the presence and absence of NiCycP (60 μM). The kinetic traces are fitted to (a) stretched exponential and (b) mono-exponential decays. | ||
A simple kinetic analysis based on 2 parallel decay pathways for RuP− in solution, namely forward electron transfer from RuP− to NiCyc which is in competition with back electron between RuP− and oxidised ascorbate, has been reported elsewhere.17 Here we apply the same model to calculate an electron transfer yield on the order of 82% for the reduction of NiCycP by photogenerated RuP− on ZrO2/RuP/NiCycP, see ESI† Section 3 for details. Experiments carried out using RuP (8.9 μM) in 0.1 M ascorbate (pH 4) solution also show the rapid formation of RuP−, observed by TA spectroscopy at 510 nm; however in contrast to the immobilised system in solution the addition of NiCycP only slightly decreases the lifetime of RuP− even at very high NiCycP concentrations (60 μM), Fig. 4b. Using the reagent concentrations employed for the solution photocatalysis experiments described above (8.9 μM RuP, 3.4 μM NiCycP) we find kapp = 1.9 × 102 s−1, leading to an estimated electron transfer yield from RuP− to NiCycP in solution of only ca. 5% indicating that the back reaction with oxidised ascorbate dominates, in line with the greatly decreased photocatalytic activity observed. Whilst the consideration of only 2 decay pathways for RuP− is a simplified model, during photocatalysis a range of alternative decay pathways are likely to become accessible as catalytic intermediates and oxidised scavengers accumulate, it does nonetheless clearly demonstrate that co-localisation of the dye and catalyst is sufficient for efficient charge transfer.
Co-immobilisation of a catalytic centre and visible light absorber onto a photochemically inert support offers a facile route to developing photocatalytic materials for CO2 reduction that is likely to be applicable to a large number of combinations of existing dyes and CO2 reduction electrocatalysts. Such an approach has been widely applied to photocatalytic water splitting20,21,27 and here we explore a model CO2 reduction system that operates in water. The focus of this communication has been on demonstrating the efficiency of the on-particle electron transfer pathway. Significantly we demonstrate that efficient on-particle electron transfer can occur, thus avoiding the need to develop complex supramolecular dye-catalyst complexes. The on-particle electron transfer pathway also offers a simple alternative to current state-of-the-art through particle dye-sensitised CO2 reduction systems30,31 that have been demonstrated in organic solvents to be highly sensitive to the presence of ionic additives. This communication also represents a significant improvement in the reported activity for light-driven CO2 reduction by NiCyc, and ZrO2/RuP/NiCycP is one of only a relatively small number of water active CO2 reduction photocatalysts. Currently our selectivity towards CO2 is relatively low, despite NiCyc being known to be a highly selective electrocatalyst. This may be in part due to the use of RuP in aqueous solutions (Fig. S19, ESI†) or the choice of electron donor and screening studies are now underway to explore alternative electron donors and lower cost sensitisers and these will be reported shortly.
The EPSRC is acknowledged for a fellowship (EP/K006851/1) supporting AJC and JJW and for equipment (EP/K031511/1) and a studentship (EP/J500471/1) for TJW. PF thanks the Royal Society for funding. The authors thank Dr V. Dhanak for XPS access.
Notes and references
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Chem. Sci., 2016, 7, 4364–4371 RSC.
- A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800–1807 CrossRef CAS PubMed.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS PubMed.
- H. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef CAS PubMed.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- G. Neri, I. M. Aldous, J. J. Walsh, L. J. Hardwick and A. J. Cowan, Chem. Sci., 2016, 7, 1521–1526 RSC.
- J. P. Petit, P. Chartier, M. Beley and J. P. Deville, J. Electroanal. Chem., 1989, 269, 267–281 CrossRef CAS.
- M. G. Bradley, T. Tysak, D. J. Graves and N. A. Viachiopoulos, J. Chem. Soc., Chem. Commun., 1983, 349 RSC.
- C. A. Craig, L. Spreer, J. W. Otvos and M. Calvin, J. Phys. Chem., 1990, 94, 7957–7960 CrossRef CAS.
- J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1987, 2105 RSC.
- M. A. Méndez, P. Voyame and H. H. Girault, Angew. Chem., Int. Ed., 2011, 50, 7391–7394 CrossRef PubMed.
- S. Montanaro, C. Herrero, D. Merli, M. Fagnoni, A. Poggi, S. Protti, S. Sheth and A. Albini, Green Process Synth., 2013, 2, 335–343 CAS.
- D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85, 1257–1305 CrossRef CAS.
- C. Herrero, A. Quaranta, S. El Ghachtouli, B. Vauzeilles, W. Leibl and A. Aukauloo, Phys. Chem. Chem. Phys., 2014, 16, 12067 RSC.
- E. Kimura, X. Bu, M. Shionoya, S. Wada and S. Maruyama, Inorg. Chem., 1992, 31, 4542–4546 CrossRef CAS.
- E. Kimura, S. Wada, M. Shionoya and Y. Okazaki, Inorg. Chem., 1994, 33, 770–778 CrossRef CAS.
- M. Hansen, F. Li, L. Sun and B. König, Chem. Sci., 2014, 5, 2683–2687 RSC.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- G. Neri, J. J. Walsh, C. Wilson, A. Reynal, J. Y. C. Lim, X. Li, A. J. P. White, N. J. Long, J. R. Durrant and A. J. Cowan, Phys. Chem. Chem. Phys., 2014, 17, 1562–1566 RSC.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- S. Füzerová, J. Kotek, I. Císařová, P. Hermann, K. Binnemans and I. Lukeš, Dalton Trans., 2005, 2908–2915 RSC.
- B. I. Lemon, F. Liu and J. T. Hupp, Coord. Chem. Rev., 2004, 248, 1225–1230 CrossRef CAS.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- A. Králík, M. Hansen and B. König, RSC Adv., 2016, 6, 5739–5744 RSC.
- G. B. Balazs and F. C. Anson, J. Electroanal. Chem., 1993, 361, 149–157 CrossRef CAS.
- H. Park, E. Bae, J. J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740–8749 CrossRef CAS PubMed.
- E.-G. Ha, J.-A. Chang, S.-M. Byun, C. Pac, D.-M. Jang, J. Park and S. O. Kang, Chem. Commun., 2014, 50, 4462 RSC.
- D.-I. Won, J.-S. Lee, J.-M. Ji, W.-J. Jung, H.-J. Son, C. Pac and S. O. Kang, J. Am. Chem. Soc., 2015, 137, 13679–13690 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, catalyst characterisation and supporting photocatalytic and transient spectroscopic measurements. CCDC 1511267. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08590c |
| This journal is © The Royal Society of Chemistry 2016 |