
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCellular delivery of enzyme-loaded DNA origami†
Ari
Ora‡
a,
Erika
Järvihaavisto‡
a,
Hongbo
Zhang
b,
Henni
Auvinen
a,
Hélder A.
Santos
 b,
Mauri A.
Kostiainen
*a and
Veikko
Linko
*a
b,
Mauri A.
Kostiainen
*a and
Veikko
Linko
*a
aBiohybrid Materials, Department of Biotechnology and Chemical Technology, Aalto University, P. O. Box 16100, FI-00076 Aalto, Finland. E-mail: mauri.kostiainen@aalto.fi; veikko.linko@aalto.fi
bDivision of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00790 Helsinki, Finland
First published on 11th November 2016
Abstract
In this communication, we show that active enzymes can be delivered into HEK293 cells in vitro when they are attached to tubular DNA origami nanostructures. We use bioluminescent enzymes as a cargo and monitor their activity from a cell lysate. The results show that the enzymes stay intact and retain their activity in the transfection process. The method is highly modular, which makes it a compelling candidate for a great variety of delivery applications.
In molecular medicine, various materials can be used in creating applicable drug delivery vehicles, such as metallic nanoparticles, viruses and polymers.1–3 However, one of the challenges is to develop carriers and advanced systems that are concurrently safe, efficient, biocompatible and entirely modular. In this respect, DNA nanotechnology offers an intriguing approach: fully programmable, custom-shaped DNA nanostructures4,5 can meet the prerequisites for smart nanocarriers, and therefore, they possess a huge potential in diverse biomedical applications.6–8
One of the most convenient techniques to build DNA nanostructures is to utilize scaffolded DNA origami,9 which facilitates robust, nanometer-level precise fabrication of arbitrary DNA shapes.10–13 Moreover, the method has served as an important starting point for developing user-friendly software for designing DNA objects14–16 and completely new design strategies for DNA-based nanoconstruction.17–20 The structural versatility provided by the DNA origami approach has yielded many intriguing applications, including optical nanodevices,21 custom-shaped metal nanoparticles,22–24 and artificial ion channels.25
Recently, there has been a growing interest towards the nanomedical implementations of the DNA nanotechnology, such as DNA sensors,26 logic-gated DNA nanorobots for controlling cell signaling27 and DNA origami structures for circumventing drug resistance, as well as delivering small anti-cancer drugs into cells both in vitro and in vivo.28–32 Other examples of DNA-based delivery systems include cages for transporting siRNA motifs33 and DNA tubes for CpG-triggered immunostimulation.34 One of the challenges in cellular transport of DNA-based nanostructures is their relatively poor transfection rates.35 However, it has been shown that the transfection can be improved by coating the structures with virus proteins36 or by introducing DNA intercalators37 that modify the surface properties of the objects. Furthermore, lipid membrane encapsulation can significantly increase the pharmacokinetic bioavailability of the DNA structures and decrease immune activation.38 Therefore, the recent developments in the field prospectively make DNA origami nanostructures as suitable candidates for smart drug delivery vehicles and carriers in advanced therapeutics.
In this communication, we show how a DNA origami loaded with active molecular components can be delivered into cells (Fig. 1). We employed bioluminescent enzymes as a cargo for the tubular DNA origami and transfected the formed complex into cells in vitro. By using these detection-sensitive enzymes, we were able to demonstrate the activity of the delivered enzymes from the cell lysate (after transfection) using a luminescence assay. The assay shows that the enzymes can stay intact in the transfection process and retain their catalytic activity, thus demonstrating the feasibility of the proposed delivery system. We believe that the system presented here can find uses, for example, in enzyme replacement therapy. Similarly, the modular DNA origami approach could be used in transporting other pivotal molecules and complexes into cells, thus enabling highly sophisticated medical treatments. Loading of the origami can be realized by attaching molecules via DNA hybridization or by using other linking techniques, such as avidin–biotin interaction. Furthermore, encapsulating active enzymes inside hollow origami cages has recently been demonstrated to efficiently protect the enzymes against proteolytic degradation.39
 | ||
| Fig. 1 A DNA origami equipped with three biotinylated binding sites (on the inner surface) is loaded with streptavidin-modified Lucia luciferase enzymes (LUC). Loaded origamis are transiently transported into cells, and subsequently, the cells are washed in order to remove free and cell membrane-bound complexes. Activity of the delivered LUC is measured from the cell lysates using coelenterazine-based luminescence assay. | ||
The DNA origami used in this work is a hexagonal tube (HT)40 (see Fig. 1), and it is equipped with three biotinylated (B) binding sites on its inner surface. The biotins are assigned for the avidin-modified cargo (three enzymes should fit in the cavity of the carrier). The DNA origami structure (hexagonal tube with biotin = HTB) was assembled as explained previously,40 and after the folding the formed structures were purified by PEG-purification41 or by spin-filtering in the 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)/NaOH buffer (6.5 mM HEPES, pH adjusted to 6.8) (see ESI†). The folded and purified HTBs were characterized using transmission electron microscopy (TEM) and agarose gel electrophoresis (see Fig. 2), which revealed proper folding of the HTBs and the efficient removal of the excess staple strands from the folding solution.
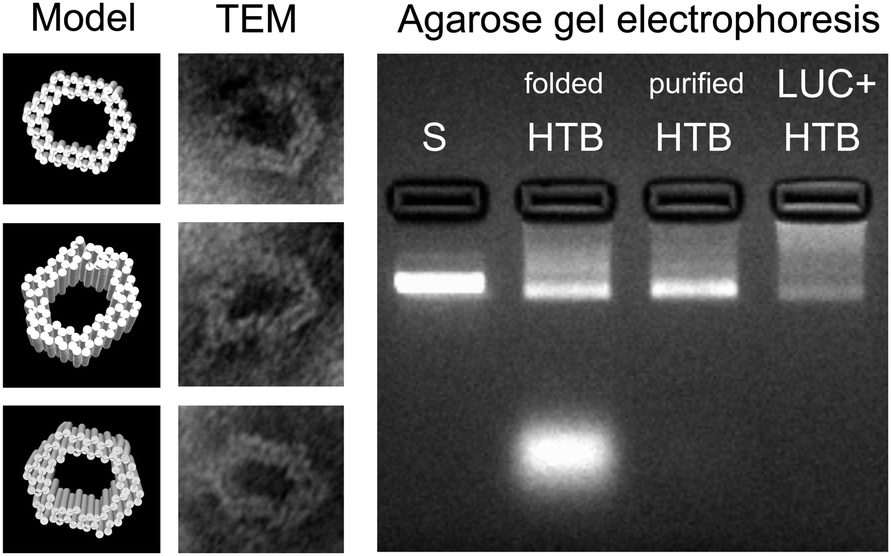 | ||
| Fig. 2 Left: CanDo-simulated14,15 solution shape of the designed DNA origami40 and TEM images of the structures with corresponding orientations. The width of the origami is between 27–33 nm and the cavity is 14–21 nm wide. Right: Agarose gel electrophoresis of the hexagonal tubes with biotin binding sites (folded and purified HTB) and hexagonal tubes with LUC-enzymes (LUC + HTB). Structures are electrophoresed after folding and after PEG-purification, and their running speeds are compared to the scaffold strand M13mp18 (S). Purification (PEG-based or spin-filtering) removes practically all the excess staple strands (bright area at the bottom of the folded HTB lane) and most of the unbound LUC-enzymes (see also ESI†). | ||
After purification, HTB origamis were incubated at least 6 h with the bioluminescent streptavidin-lucia (LUC) luciferase enzymes (InvivoGen). The enzymes were added in excess amounts, and the unbound enzymes were removed again by PEG-purification or by spin-filtering (HEPES/NaOH buffer) (see ESI†). HTB origamis with LUC-enzymes (LUC + HTB) were also analyzed by gel electrophoresis (Fig. 2). Addition of LUC smears the origami band, thus indicating complexation with origamis (see the LUC + HTB lane in Fig. 2). After the structural characterization and before the transfection, the bioluminescence decay kinetics of the fabricated LUC + HTB, free luciferase (LUC (adjusted)) and bare origami (HTB) were analyzed (see Fig. 3). The luminescence assay and analysis are carried out similarly as explained previously;42 briefly, the activity of each sample was characterized by mixing 10 μl of the sample solution with the 50 μl of commercial coelenterazine-based luminescence assay reagent (QUANTI-Luc, InvivoGen), and by monitoring the luminescence by luminometer (BioTek Cytation 3). The resulted luminescence decay as a function of time is shown in Fig. 3. The concentration of the free luciferase sample (LUC (adjusted)) was titrated to match the luminescence decay curve of LUC + HTB sample. Typically, the concentration of free luciferase (LUC (adjusted)) was 150–600 nM, i.e., 5–15 times the origami concentration (varied between 10–100 nM). Therefore, the results indicate that LUC binds not only to biotinylated strands, but also unspecifically to DNA origami, and that LUC + HTB sample may contain a modest amount of enzymes that were not removed in the purification process (see ESI†). In addition, the activity of enzymes that are attached to DNA origami have been found to be higher than that of free enzymes,39 which could also explain the effect. However, the free LUC sample (used as a control in the transfection studies) was always adjusted to match LUC + HTB kinetics before the transfection in order to compensate these effects.
 | ||
| Fig. 3 Enzyme activity and the sample quality were verified before transfection by the luminescence decay assay. The graph shows typical luminescence decay as a function of time for a purified DNA origami loaded with luciferases (LUC + HTB), similar amount of free luciferase (LUC (adjusted)) and an empty DNA origami (HTB). The concentration in the free luciferase sample is adjusted to match the decay curve of the LUC + HTB sample. The first data points were recorded 10 seconds after adding the coelenterazine-based luminescence assay reagent to the origami solutions. The inset depicts the statistics of the luminescence assay (relative luminescence intensity): the maximum luminescence intensity is depicted with 100% in each measurement and this value was compared to the maximum intensity of the corresponding LUC and HTB samples. | ||
Fully characterized and tested samples (LUC + HTB, LUC (adjusted) and HTB at ∼1 nM concentration) were then transfected into HEK293 cells transiently (see ESI† for details of transfection and cell culturing). To visualize the different compartments under confocal microscopy, we labeled cell membrane with Cell Mask Deep Red, HTB origamis with fluorescent Cy3 dyes (five Cy3-modified strands per each origami) and LUC with Pacific Blue dye (labeled with Molecular Probes' Pacific Blue™ Protein Labeling Kit) and followed the transfection. The viability of cells was always verified before imaging. After 12 h of transfection, it was observed that the origamis and enzymes were found in cells (Fig. 4A and B). The results show that in some cases LUC and HTB were clearly co-localized (Fig. 4A), but it also seems that unspecifically bound LUC might detach from the HTB in the transfection (see ESI†). Moreover, bare LUC (LUC (adj.)) was transfected to some extent (Fig. 4B). Furthermore, we also tested longer (36 h) transfection, which showed that HTB is co-localized with the cell nuclei (see ESI†).
 | ||
| Fig. 4 (A and B) Confocal micrographs of LUC + HTB (A) and LUC (adjusted) without HTB (B) samples after 12 h transfection without PEI: LUC is labeled with Pacific Blue dye (blue), HTB with Cy3-modified strands (green) and cell membrane by cell mask deep red (red). (C) The statistics of the luminescence assay (relative luminescence intensity) measured from the cell lysate (without PEI): the maximum luminescence intensity corresponds 100% in each sample set (LUC + HTB sample showed always the highest activity, and this value was compared to the maximum intensity of the corresponding LUC (adj.) and HTB samples). | ||
For monitoring the transfection (12 h) and the activity of the samples via luminescence assay, the cells were washed carefully 3 times with PBS in order to remove the complexes that were possibly attached to cell membranes or the cell culturing plates. Cells were lysed using coelenterazine-based substrate and the bioluminescence was immediately detected as explained above. The luminescence decay of the samples was significantly slower in cell lysates than before transfection, presumably due to the dilution of the samples during the transfection. The slower kinetics enabled detection of clear differences between the luminescence intensity levels of each sample within the time-scale of several minutes. Fig. 4C shows the normalized maximum luminescence intensities (the maximum intensity in each measurement represents 100% relative luminescence) for the purified LUC + HTB, LUC (adj.) and bare HTB samples. The results show that although the activity of LUC + HTB and bare LUC were adjusted to the same level before transfection (Fig. 3 inset), the LUC + HTB displays roughly 3 times as high intensity as the free LUC sample after the transfection (Fig. 4C).
As additional control experiments, we used only the structural components of the DNA origami (short DNA strands, i.e., staples, and a long scaffold strand), which we incubated with LUC and studied how these DNA strands alone could affect the results. With the purification methods used here, it is impossible to separate staples and scaffold strands from the excess and unbound LUC. Nevertheless, we then used the unpurified samples (containing equal amounts of LUC in each sample, i.e., 15 times HTB concentration). It was observed that the luminescence intensity levels were enhanced using the short DNA staples or the scaffold strand alone. As a result, the luminescence level of the LUC + staples sample was similar to LUC + HTB but higher than LUC + scaffold and clearly higher than bare LUC. All the abovementioned experiments were performed without any transfection reagent, but as a control, we also carried out the experiments with a common transfection reagent polyethylenimine (PEI, ∼25 kDa, linear). As expected, PEI can enhance the transfection. The data from the controls and the samples transfected with PEI are presented in ESI.† Overall, all the results prove the feasibility of the proposed method, since the DNA origami working concentrations could be as low as 500 pM.
In conclusion, we have demonstrated how a designer DNA origami loaded with functional cargo (in principle any streptavidin- or biotin-modified cargo) can be transported into cells. In general, the cellular delivery of functional proteins is essential in molecular medicine, therapeutics and nanomedical engineering.43–49 Here, the proof-of-principle delivery works with or without the transfection reagent and without any surface functionalization. Therefore, we strongly believe that the transfection properties could be significantly enhanced by introducing specified cell targeting ligands, surface modulations and by further taking advantage of the structural modularity of DNA origami.36–38,50 Recently, it was shown that DNA-coated enzymes can transfect efficiently and retain their activity in cells.51 Further, encapsulation or anchoring enzymes using DNA origami structures could protect enzymes against protease digestion,39 which can lead to interesting implementations in nanomedicine. Recently, it has also been demonstrated that reaction rates of the enzyme-loaded origamis can be controlled by coating the origamis with synthetic polymers.42 Thus, this work further intensifies the development of smart and tailored nanocarrier systems that are able to facilitate a plethora of biomedical applications in the near future.
We acknowledge the Academy of Finland (263504, 267497, 273645, 286845, 252215, 281300), Biocentrum Helsinki, Emil Aaltonen Foundation, Erkko Foundation (4704010), University of Helsinki Research Funds, and the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013, grant no. 310892). The work was carried out under the Academy of Finland's Centres of Excellence Programme (2014–2019) and made use of the Aalto Nanomicroscopy Centre and Meilahti Clinical & Basic Proteomics, Core Facility, Biomedicum-Helsinki premises.
References
- A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS PubMed.
- Y. Ma, R. J. M. Nolte and J. J. L. M. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS PubMed.
- N. Nishiyama and K. Kataoka, Pharmacol. Ther., 2006, 112, 630–648 CrossRef CAS PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- V. Linko and H. Dietz, Curr. Opin. Biotechnol., 2013, 24, 555–561 CrossRef CAS PubMed.
- F. Kong, X. Zhang, H. Zhang, X. Qu, D. Chen, M. Servos, E. Mäkilä, J. Salonen, H. A. Santos, M. Hai and D. A. Weitz, Adv. Funct. Mater., 2015, 25, 3330–3340 CrossRef CAS.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- D.-N. Kim, F. Kilchherr, H. Dietz and M. Bathe, Nucleic Acids Res., 2012, 40, 2862–2868 CrossRef CAS PubMed.
- K. Pan, D.-N. Kim, F. Zhang, M. R. Adendorff, H. Yan and M. Bathe, Nat. Commun., 2014, 5, 5578 CrossRef CAS PubMed.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed.
- D. Han, S. Pal, Y. Yang, S. Jiang, J. Nangreave, Y. Liu and H. Yan, Science, 2013, 339, 1412–1415 CrossRef CAS PubMed.
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Högberg, Nature, 2015, 523, 441–444 CrossRef CAS PubMed.
- R. Veneziano, S. Ratanalert, K. Zhang, F. Zhang, H. Yan, W. Chiu and M. Bathe, Science, 2016, 352, 1534 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- S. Helmi, C. Ziegler, D. J. Kauert and R. Seidel, Nano Lett., 2014, 14, 6693–6698 CrossRef CAS PubMed.
- W. Sun, E. Boulais, Y. Hakobyan, W. L. Wang, A. Guan, M. Bathe and P. Yin, Science, 2014, 346, 1258361 CrossRef PubMed.
- B. Shen, V. Linko, K. Tapio, M. A. Kostiainen and J. J. Toppari, Nanoscale, 2015, 7, 11267–11272 RSC.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- S. Ranallo, A. Amodio, A. Idili, A. Porchetta and F. Ricci, Chem. Sci., 2016, 7, 66–71 RSC.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 861–864 CrossRef PubMed.
- Q. Jiang, C. Song, J. Nangreave, X. Liu, L. Lin, D. Qiu, Z.-G. Wang, G. Zou, X. Liang, H. Yan and B. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS PubMed.
- Y.-X. Zhao, A. Shaw, X. Zeng, E. Benson, A. M. Nyström and B. Högberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- M. Fu, L. Dai, Q. Jiang, Y. Tang, X. Zhang, B. Ding and J. Li, Chem. Commun., 2016, 52, 9240–9242 RSC.
- P. D. Halley, C. R. Lucas, E. M. McWilliams, M. J. Webber, R. A. Patton, C. Kural, D. M. Lucas, J. C. Byrd and C. E. Castro, Small, 2016, 12, 308–320 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- A. H. Okholm, J. S. Nielsen, M. Vinther, R. S. Sørensen, D. Schaffert and J. Kjems, Methods, 2014, 67, 193–197 CrossRef CAS PubMed.
- J. Mikkilä, A.-P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- J. Brglez, P. Nikolov, A. Angelin and C. M. Niemeyer, Chem. – Eur. J., 2015, 21, 9440–9446 CrossRef CAS PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- V. Linko, M. Eerikäinen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351–5354 RSC.
- E. Stahl, T. G. Martin, F. Praetorius and H. Dietz, Angew. Chem., Int. Ed., 2014, 126, 12949–12954 CrossRef.
- J. K. Kiviaho, V. Linko, A. Ora, T. Tiainen, E. Järvihaavisto, J. Mikkilä, H. Tenhu, Nonappa and M. A. Kostiainen, Nanoscale, 2016, 8, 11674–11680 RSC.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- F. C. Simmel, Curr. Opin. Biotechnol., 2012, 23, 516–521 CrossRef CAS PubMed.
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS PubMed.
- A. A. Eltoukhy, D. Chen, O. Veiseh, J. M. Pelet, H. Yin, Y. Dong and D. G. Anderson, Biomaterials, 2014, 35, 6454–6461 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- V. Linko, S. Nummelin, L. Aarnos, K. Tapio, J. J. Toppari and M. A. Kostiainen, Nanomaterials, 2016, 6, 139 CrossRef.
- D. H. Schaffert, A. H. Okholm, R. S. Sørensen, J. Nielsen, T. Tørring, C. B. Rosen, A. L. B. Kodal, M. R. Mortensen, K. V. Gothelf and J. Kjems, Small, 2016, 12, 2634–2640 CrossRef CAS PubMed.
- J. D. Brodin, A. J. Sprangers, J. R. McMillan and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 14838–14841 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of DNA origami preparation and purification methods. Details of cell culturing, transfection, TEM imaging, gel electrophoresis, and confocal microscopy. Additional confocal images. Luminescence assay for free enzymes and spin-filtered enzymes. A list of DNA origami strands. See DOI: 10.1039/c6cc08197e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |