
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic synthesis of natural (+)-aristolochene from a non-natural substrate†
Juan A.
Faraldos
a,
Daniel J.
Grundy
a,
Oscar
Cascon
a,
Stefano
Leoni
a,
Marc W.
van der Kamp
 b and
Rudolf K.
Allemann
*a
b and
Rudolf K.
Allemann
*a
aSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 3AT, UK. E-mail: AllemannRK@cardiff.ac.uk
bSchool of Biochemistry, Faculty of Biomedical Sciences & Centre for Computational Chemistry, School of Chemistry, University of Bristol, Biomedical Sciences Building, University Walk, Bristol BS8 1TD, UK
First published on 3rd November 2016
Abstract
The sesquiterpene cyclase aristolochene synthase from Penicillium roquefortii (PR-AS) has evolved to catalyse with high specificity (92%) the conversion of farnesyl diphosphate (FDP) to the bicyclic hydrocarbon (+)-aristolochene, the natural precursor of several fungal toxins. Here we report that PR-AS converts the unnatural FDP isomer 7-methylene farnesyl diphosphate to (+)-aristolochene via the intermediate 7-methylene germacrene A. Within the confined space of the enzyme's active site, PR-AS stabilises the reactive conformers of germacrene A and 7-methylene germacrene A, respectively, which are protonated by the same active site acid (most likely HOPPi) to yield the shared natural bicyclic intermediate eudesmane cation, from which (+)-aristolochene is then generated.
Terpenoids are found in all forms of life and comprise one of the largest and most structurally diverse families of natural products.1–4 Sesquiterpenoids are biosynthesized from the universal linear C15-precursor farnesyl diphosphate (FDP, 1), which is converted in electrophilic cyclization reactions of often exquisite specificity to >300 mostly cyclic hydrocarbon products in class I sesquiterpene synthase catalysed processes.5 The electrophilic nature and stereochemical precision of these cascade reactions have been probed in vitro with FDP isotopologues6–11 as well as with substituted FDPs that carry fluoro,12–16 methyl17,18 and phenyl substituents.19 The ability of sesquiterpene synthases to bind such synthetic FDP analogues as competitive inhibitors13,19,20 and occasionally as substrates11,12,14,16–18 demonstrates their plasticity and catalytic flexibility, properties that have been exploited for the synthesis of several complex unnatural molecules.12,14,17,21
Aristolochene synthase from Penicillium roquefortii (PR-AS) efficiently (92%) catalyses the cyclisation of 1 to the bicyclic eremophilane hydrocarbon (+)-aristolochene (2), the precursor of several highly oxygenated mycotoxins such us PR-toxin.8,22 This cyclisation, which generates two 6-membered rings and introduces two double bonds and three stereo-centres with high specificity,23–25 goes through the neutral intermediate germacrene A (3), a flexible cyclodecadiene hydrocarbon that undergoes reprotonation to eudesmane cation (4),24,25 most likely by proton transfer from the by-product diphosphate (HOPPi).26–28 Rearrangement of 4 by way of a hydride and a methyl shift is followed by a selective deprotonation at C8 to yield (+)-aristolochene (2) (Scheme 1) together with a small amount of ∼1% valencene (5), the double bond isomer of 2 that results from deprotonation at C6 rather than C8. PR-AS also releases ∼7% germacrene A (3) prior to protonation of the central C6,C7-double bond of 3, possibly as a consequence of a conformational change prior to eudesmane cation formation.24,25
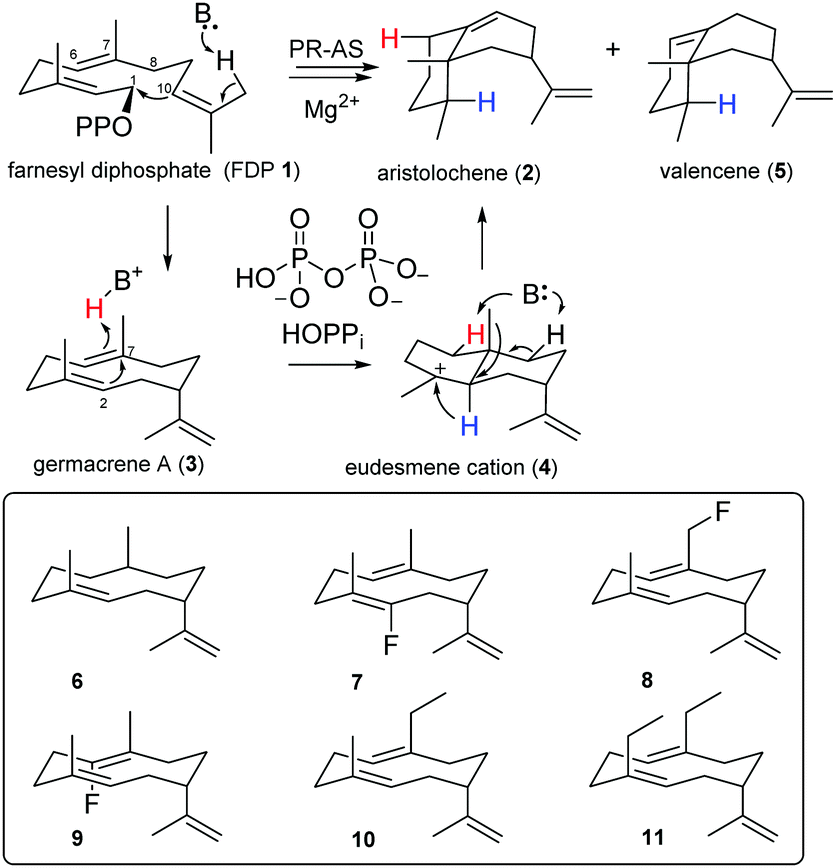 | ||
| Scheme 1 Biosynthesis of (+)-aristolochene (2) and unnatural germacrenoids (6–11) generated by PR-AS from the corresponding FDP analogues. | ||
The intermediacy of eudesmane cation (4) was inferred from site directed mutagenesis experiments29,30 and inhibition studies with ammonium and iminium salt analogues of 4.31,32 Evidence for the formation of germacrene A (3) was first obtained when PR-AS was challenged with 6,7-dihydroFDP, which was turned over to the corresponding dihydrogermacrene A (6).21 Similarly, the PR-AS catalysed turnover of several fluoro- and methyl-substituted FDP analogues to the corresponding altered germacrene A analogues (6–11)12,14,17,21 illustrated the synthetic potential of sesquiterpene synthases for organic synthesis.11,14,16–18
If HOPPi protonates germacrene A (3) and its corresponding base (−OPPi) acts to abstract a proton from eudesmane cation (4) in the final step of PR-AS catalysis27,28 (Scheme 1), the synthetic space accessible to PR-AS might potentially be expandable and 7-methylene FDP (12), an unnatural FDP isomer that the enzyme will never be exposed to in the host organism, could be efficiently converted to aristolochene by reprotonation of the C7,C14 double bond of the intermediate 7-methylene germacrene A (13) to yield eudesmane cation (4) (Scheme 2), accomplishing for the first time a one-step enzymatic synthesis of a structurally complex natural sesquiterpene (2) from a non-natural, synthetic precursor. Comparison of the relative reaction rates of the hydration for a series of simple olefins in aqueous sulfuric acid33 suggests that both 3 and 13 should have similar reactivity towards protonation under general acid catalysis.
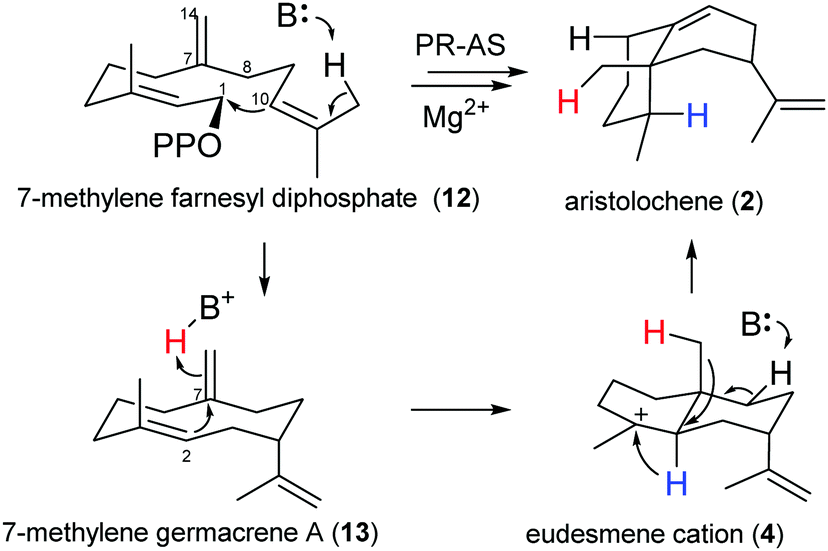 | ||
| Scheme 2 The PR-AS catalysed conversion of 7-methylene-FDP (12) to (+)-aristolochene (2). | ||
To assess the feasibility of the PR-AS catalysed cyclisation of 7-methylene-FDP (12) to 2, quantum mechanical geometry optimisation and docking into the enzyme active were performed. Previously, it was suggested that 3 exists as a mixture of three conformers in solution with the UU conformer (enforced by PR-AS, Fig. 1) as the predominant form (52%).34 Molecular modeling, in which the hydrocarbon chain was confined to the approximate active site volume, confirmed that like for 3, the most stable conformation of 13 is the UU form. Upon relaxation in vacuo, distances between C2 and C7 in 3 and 13 are ∼3.3 Å. When the volume accessible to 3 and 13 is constrained, marked shortening of the C2–C7 distances were observed to less than 2.5 Å (Fig. 1). Docking of the constrained structures of 3 and 13 in the (highly homologous) closed active site of aristolochene synthase from A. terreus35 (Fig. 2) demonstrates that both fit in the active site pocket in similar conformations and in an orientation consistent with the expected reaction path, with e.g.−OPPi abstracting a proton from 1 or 12. As an indicator of the direction of the second ring closure reaction (Scheme 1), we modeled the LUMOs of 3 and 13 both in vacuo and in the enzyme environment. Upon constrained optimisation of the vacuum relaxed structures, the nodal configurations markedly changed for both 3 and 13, and they remain similar when the enzyme environment is taken into account. The calculated LUMOs highlight the favourable orbital overlap across the ring and demonstrate the feasibility of the acid-catalysed C2,C7 ring closure for both 3 and 13 inside PR-AS (Fig. 1).
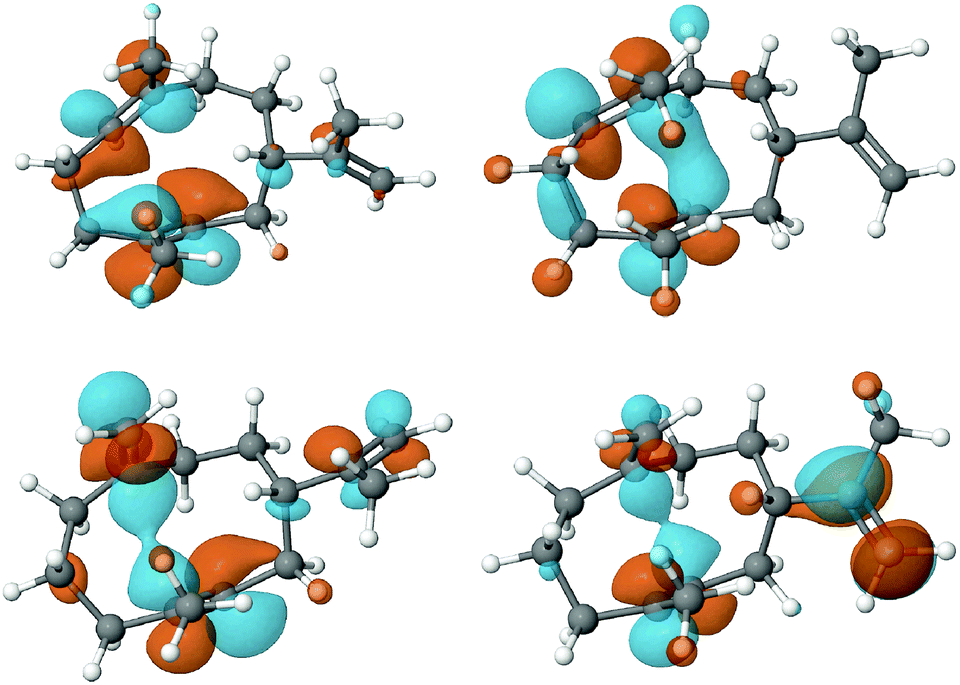 | ||
| Fig. 1 Geometries and LUMOs of 3 (above) and 13 (below) in their reactive UU conformations, after unconstrained optimisation (vacuum) (left) and constrained optimisation and docking into the enzyme active site (right). Under constrained volume conditions 3 and 13 become more similar and the 2,7-contact shorter than the 2,6 contact (see also Fig. S2, ESI†). | ||
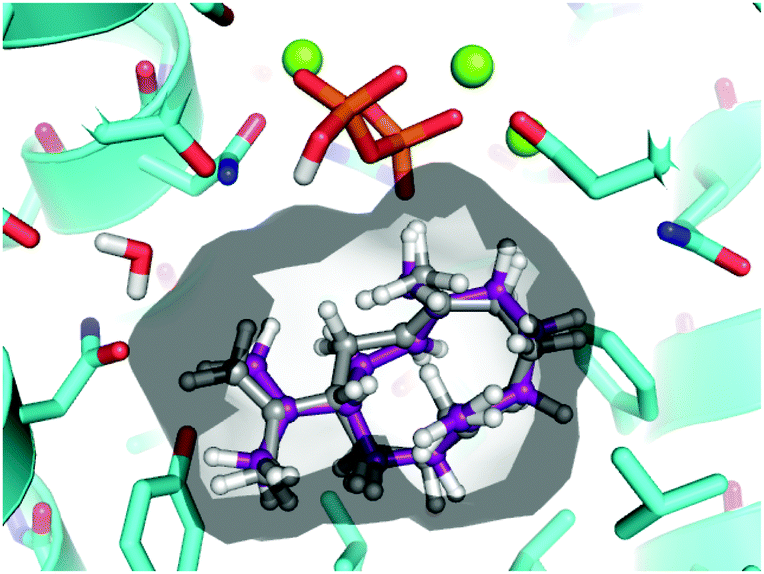 | ||
| Fig. 2 Constrained geometries of 3 (grey carbons) and 13 (purple carbons) as docked in the active site of aristolochene synthase from A. terreus (cyan carbons; PDB ID: 4KVY, chain A). The solvent accessible surface of the enzyme active site is shown. For clarity, hydrogen atoms are removed from protein residues. | ||
After expressing the PR-AS cDNA in E. coli (ESI†), GC-MS analysis of incubations of 7-methylene FDP (12) with purified recombinant PR-AS revealed the in vitro formation of a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture of two major products (Fig. 3). Comparison of the elution profiles in the gas chromatograms and MS fragmentation patterns from parallel incubations of 1 and 12 with PR-AS and Solidago canadensis germacrene A synthase (GAS)36 indicated that the two products generated by PR-AS from 12 were aristolochene (2) (50.1%, 15.24 minutes) and the germacrene A analogue 13 (30.1%, 16.27 minutes). In addition to 2 and 13 three minor products were formed (Fig. 3) that made up 19.8% of the total amount of products. Their identity was not unambiguously determined but the possibility that they were linear 7-methylene farnesenes was ruled out by comparison of the GC chromatograms and MS spectra of the products obtained from the reaction of 12 with Mentha X piperita (E)-β-farnesene synthase (EBFS).37 The GC retention times and comparison with the MS of the products formed from incubations of 1 with PR-AS suggested that the compound that eluted at 15.55 (ca. 10%) min was valencene (5) (Fig. 3). The production of 2 from diphosphate 12 suggests that both reactions go via eudesmane cation (4) (Schemes 1 and 2). In the case of 13, the exo double bond may be positioned slightly less favourably for protonation so that 13 is longer lived in the active site than 3 and more germacrenoid 13 (30%) is released than is the case for 3 (7%). Alternatively, 13 might bind less tightly to the active site of PR-AS. Nevertheless, the similarity of 1 and 12 and the plasticity of PR-AS lead to similar active site conformations for both substrates so that the initial 1,10-ring closure reactions to 3 and 13 occur along closely related reaction paths. The molecular modeling results demonstrate the importance of constraining the germacrene A intermediates 3 and 13 in the active site for the protonation of the endo-C6,C7 and exo-C7,C14 double bonds.
:3 mixture of two major products (Fig. 3). Comparison of the elution profiles in the gas chromatograms and MS fragmentation patterns from parallel incubations of 1 and 12 with PR-AS and Solidago canadensis germacrene A synthase (GAS)36 indicated that the two products generated by PR-AS from 12 were aristolochene (2) (50.1%, 15.24 minutes) and the germacrene A analogue 13 (30.1%, 16.27 minutes). In addition to 2 and 13 three minor products were formed (Fig. 3) that made up 19.8% of the total amount of products. Their identity was not unambiguously determined but the possibility that they were linear 7-methylene farnesenes was ruled out by comparison of the GC chromatograms and MS spectra of the products obtained from the reaction of 12 with Mentha X piperita (E)-β-farnesene synthase (EBFS).37 The GC retention times and comparison with the MS of the products formed from incubations of 1 with PR-AS suggested that the compound that eluted at 15.55 (ca. 10%) min was valencene (5) (Fig. 3). The production of 2 from diphosphate 12 suggests that both reactions go via eudesmane cation (4) (Schemes 1 and 2). In the case of 13, the exo double bond may be positioned slightly less favourably for protonation so that 13 is longer lived in the active site than 3 and more germacrenoid 13 (30%) is released than is the case for 3 (7%). Alternatively, 13 might bind less tightly to the active site of PR-AS. Nevertheless, the similarity of 1 and 12 and the plasticity of PR-AS lead to similar active site conformations for both substrates so that the initial 1,10-ring closure reactions to 3 and 13 occur along closely related reaction paths. The molecular modeling results demonstrate the importance of constraining the germacrene A intermediates 3 and 13 in the active site for the protonation of the endo-C6,C7 and exo-C7,C14 double bonds.
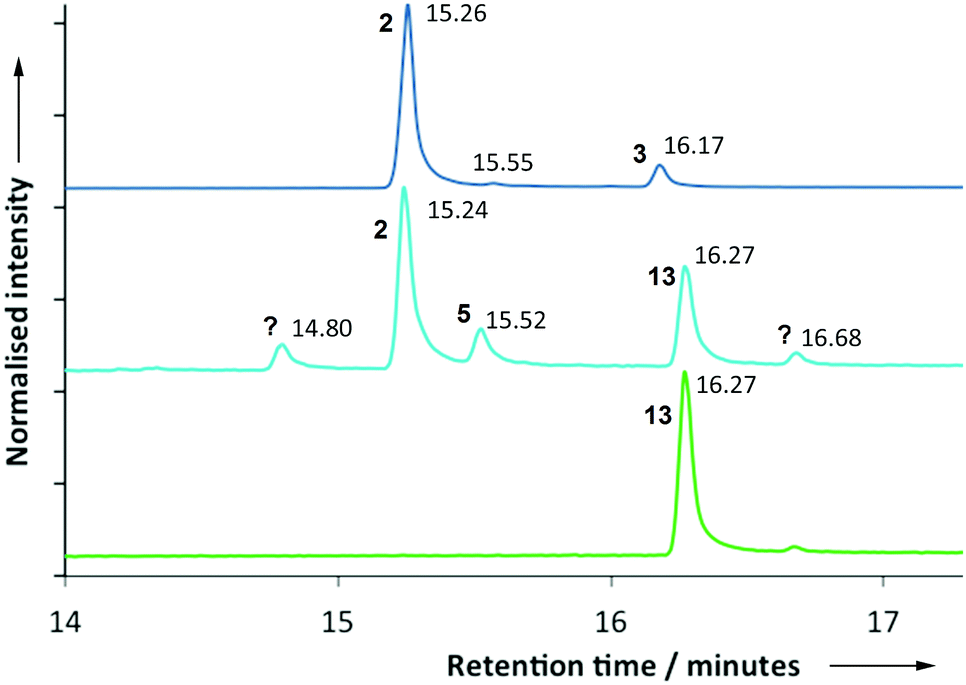 | ||
| Fig. 3 Gas chromatographic analyses of the products produced from incubations of PR-AS with FDP (1) (blue, top) and 7-methylene FDP (12) (light blue, middle). For comparison, the result from an incubation of germacrene A synthase (GAS) with 7-methylene FDP (12) is also shown (green, bottom). | ||
The investigation reported here demonstrates the remarkable versatility of PR-AS, which effectively turns over an unnatural synthetic precursor to the natural products aristolochene (2) and valencene (5). The PR-AS catalysed conversion of the unnatural substrate 12 to (+)-aristolochene (2) illustrates the power of substrate engineering for the in vitro synthesis of structurally complex natural products utilizing sesquiterpene synthases as biocatalysts. Our in silico work also highlights the importance of constraining the reactants for the reactivity of intermediates towards the generation of complex terpene products, an effect that has first been seen in the relatively easy, albeit less specific biomimetic cyclisation reactions of squalene in the absence of enzyme, where the hydrophobic effect leads to restrictions of the conformational space accessible to the hydrocarbon chain.38 For shorter chain isoprenoid diphosphates such as geranyl diphosphate, farnesyl diphosphate and geranylgeranyl diphosphate on the other hand, the enzyme is needed to act as a folding template for efficient product formation.
This work was supported by the UK's Biotechnology and Biological Sciences Research Council (BB/H01683X/1, BB/G003572/1 and BB/M026280/1), the UK's Engineering and Physical Sciences Research Council (EP/L027240/1) and the Cardiff Synthetic Biology Initiative, Cardiff University. We thank Dr Rob Jenkins, Robin Hicks, Simon Waller and Thomas Williams (Cardiff University) for assistance with mass spectrometry and NMR, and Dr Veronica Gonzalez (Cardiff University) for the preparation of GAS and EBFS. Computational resources of ARCCA at Cardiff are acknowledged.
Notes and references
- J. Gershenzon and N. Dudareva, Nat. Chem. Biol., 2007, 3, 408 CrossRef CAS PubMed.
- J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman and Hall, London, 1991 Search PubMed.
- D. E. Cane, Chem. Rev., 1990, 90, 1089 CrossRef CAS.
- D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS PubMed.
- D. J. Miller and R. K. Allemann, Nat. Prod. Rep., 2012, 29, 60 RSC.
- D. E. Cane, S. Swanson and P. P. N. Murthy, J. Am. Chem. Soc., 1981, 103, 2136 CrossRef CAS.
- D. E. Cane, H.-J. Ha, C. Pargellis, F. Waldmeier, S. Swanson and P. P. N. Murthy, Bioorg. Chem., 1985, 13, 246 CrossRef CAS.
- D. E. Cane, P. C. Prabhakaran, E. J. Salaski, P. H. M. Harrison, H. Noguchi and B. J. Rawlings, J. Am. Chem. Soc., 1989, 111, 8914 CrossRef CAS.
- C. O. Schmidt, H. J. Bouwmeester, S. Franke and W. A. König, Chirality, 1999, 362, 353 CrossRef.
- S. Picaud, P. Mercke, X. He, O. Sterner, M. Brodelius, D. E. Cane and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 448, 150 CrossRef CAS PubMed.
- J. A. Faraldos, D. J. Miller, V. Gonzalez, Z. Yoosuf-Aly, O. Cascon, A. Li and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 5900 CrossRef CAS PubMed.
- D. J. Miller, F. L. Yu, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962 CAS.
- F. Yu, D. J. Miller and R. K. Allemann, Chem. Commun., 2007, 4155 RSC.
- D. J. Miller, F. L. Yu and R. K. Allemann, ChemBioChem, 2007, 8, 1819 CrossRef CAS PubMed.
- J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess Jr., L. Smentek, R. M. Coates and P. E. O’Maille, ACS Chem. Biol., 2010, 5, 377 CrossRef CAS PubMed.
- J. A. Faraldos, Y. Zhao, P. E. O’Maille, J. P. Noel and R. M. Coates, ChemBioChem, 2007, 8, 1826 CrossRef CAS PubMed.
- O. Cascón, S. Touchet, D. J. Miller, V. Gonzalez, J. A. Faraldos and R. K. Allemann, Chem. Commun., 2012, 48, 9702 RSC.
- S. Touchet, K. Chamberlain, C. M. Woodcock, D. J. Miller, M. A. Birkett, J. A. Pickett and R. K. Allemann, Chem. Commun., 2015, 51, 7550 RSC.
- D. J. Miller, F. L. Yu, N. J. Young and R. K. Allemann, Org. Biomol. Chem., 2007, 5, 3287 CAS.
- D. E. Cane and C. Bryant, J. Am. Chem. Soc., 1994, 116, 12063 CrossRef CAS.
- D. E. Cane and Y. S. Tsantrizos, J. Am. Chem. Soc., 1996, 118, 10037 CrossRef CAS.
- D. E. Cane, Z. Wu, R. H. Proctor and T. M. Hohn, Arch. Biochem. Biophys., 1993, 304, 415 CrossRef CAS PubMed.
- D. E. Cane, P. C. Prabhakaran, J. S. Oliver and D. B. McIlwaine, J. Am. Chem. Soc., 1990, 112, 3209 CrossRef CAS.
- M. J. Calvert, P. R. Ashton and R. K. Allemann, J. Am. Chem. Soc., 2002, 124, 11636 CrossRef CAS PubMed.
- B. Felicetti and D. Cane, J. Am. Chem. Soc., 2004, 126, 7212 CrossRef CAS PubMed.
- E. Shishova, L. Di Costanzo, D. E. Cane and D. W. Christianson, Biochemistry, 2007, 46, 1941 CrossRef CAS PubMed.
- J. A. Faraldos, V. Gonzalez and R. K. Allemann, Chem. Commun., 2012, 48, 3230 RSC.
- T. A. Pemberton and D. W. Christianson, J. Antibiot., 2016, 69, 486 CrossRef CAS PubMed.
- A. Deligeorgopoulou, S. E. Taylor, S. Forcat and R. K. Allemann, Chem. Commun., 2003, 2162 RSC.
- J. A. Faraldos, A. K. Antonczak, V. González, R. Fullerton, E. M. Tippmann and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 13906 CrossRef CAS PubMed.
- J. A. Faraldos, B. Kariuki and R. K. Allemann, J. Org. Chem., 2010, 75, 1119 CrossRef CAS PubMed.
- J. A. Faraldos and R. K. Allemann, Org. Lett., 2011, 13, 1202–1205 CrossRef CAS PubMed.
- V. J. Nowlan and T. T. Tidwell, Acc. Chem. Res., 1977, 10, 252 CrossRef CAS.
- J. A. Faraldos, S. Wu, J. Chappell and R. M. Coates, Tetrahedron, 2007, 63, 7733 CrossRef CAS PubMed.
- M. Chen, N. Al-lami, M. Janvier, E. L. D'Antonio, J. A. Faraldos, D. E. Cane, R. K. Allemann and D. W. Christianson, Biochemistry, 2013, 52, 5441 CrossRef CAS PubMed.
- V. Gonzalez, S. Touchet, D. J. Grundy, J. A. Faraldos and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 14505 CrossRef CAS PubMed.
- J. A. Faraldos, V. Gonzalez, A. Li, F. Yu, M. Köksal, D. W. Christianson and R. K. Allemann, J. Am. Chem. Soc., 2012, 134, 20844 CrossRef CAS PubMed.
- T. D. Zahn, M. Eilers, Z. Guo, M. B. Ksebati, M. Simon, J. D. Scholten, S. O. Smith and R. A. Gibbs, J. Am. Chem. Soc., 2000, 122, 7135 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of FDP and 7-methylene-FDP, gas chromatograms, MS spectra, 1H-NMR spectra and molecular calculations. See DOI: 10.1039/c6cc08164a |
| This journal is © The Royal Society of Chemistry 2016 |