
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUranium(III) and thorium(IV) alkyl complexes as potential starting materials†
Andrew C.
Behrle‡
a,
Alexander J.
Myers‡
a,
Pokpong
Rungthanaphatsophon
a,
Wayne W.
Lukens
b,
Charles L.
Barnes
a and
Justin R.
Walensky
*a
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: WalenskyJ@missouri.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 16th November 2016
Abstract
The synthesis and characterisation of a rare U(III) alkyl complex, U[η4-Me2NC(H)C6H5]3, using the dimethylbenzylamine (DMBA) ligand has been accomplished. While attempting to prepare the U(IV) compound, reduction to the U(III) complex occurred. In the analogous Th(IV) system, C–H bond activation of a methyl group of one dimethylamine was observed yielding Th[η4-Me2NC(H)C6H5]2[η5-(CH2)MeNC(H)C6H5] with a dianionic DMBA ligand. The utility of these complexes as starting materials has been analyzed using a bulky dithiocarboxylate ligand to yield tetravalent actinide species.
During the Manhattan project, actinide alkyl complexes were desirable for their potential as volatile compounds for separations, especially uranium enrichment.1 More recently, organoactinide chemistry has experienced increased attention as exemplified by the Hayton and Bart groups. For example, Hayton has reported homoleptic U(IV),2 U(V), and U(VI) alkyl3 complexes as well as Th(IV) alkyl1 and aryl4 complexes while Bart has produced a series of U(IV) benzyl compounds.5,6 Nevertheless, Th(IV) and U(III) alkyl complexes7–11 remain scarce.
Recently, the Hayton group has used the lithium salt of dimethylbenzylamine (DMBA) to synthesize Th(IV) and U(IV) complexes.12,13 The lithiation of dimethylbenzylamine produces an ortho-metalated phenyl anion. This salt may be converted to the benzyl anion by reaction with potassium tert-butoxide,14,15 which is accompanied by a proton migration from the alpha-position of the benzyl methylene to the ortho-position of the phenyl. The only known complexes using this ligand transfer agent as starting material are a zirconium complex16 as well as most of the lanthanide series.15 Since the Ln(III) complexes are stabilized by this ligand, we surmised that U(III) would be stabilized in a similar fashion.
Reaction of UI3(THF)4 with three equivalents of K[Me2NC(H)C6H5] in Et2O for 3 h at −25 °C, eqn (1), results in a dark coloured solution. X-ray quality crystals were grown from a saturated toluene solution at −25 °C, and diffraction revealed the U(III) complex, U[η4-Me2NC(H)C6H5]3, 1, Fig. 1. Reaction with UCl4 also produced 1 along with half an equivalent of 1,2-bis(dimethylamino)-1,2-diphenylethane. The 1H NMR spectrum of 1 is fluxional at room temperature, but cooling to −78 °C made the spectrum assignable. The 1H NMR spectrum is paramagnetically shifted, and the amine methyl resonances are inequivalent at 47 ppm and −71 ppm. The methine proton is located at −94 ppm. Complex 1 is thermally unstable above room temperature but stable when stored cold in the solid-state. As mentioned previously, this compound represents a rare U(III) alkyl complex.
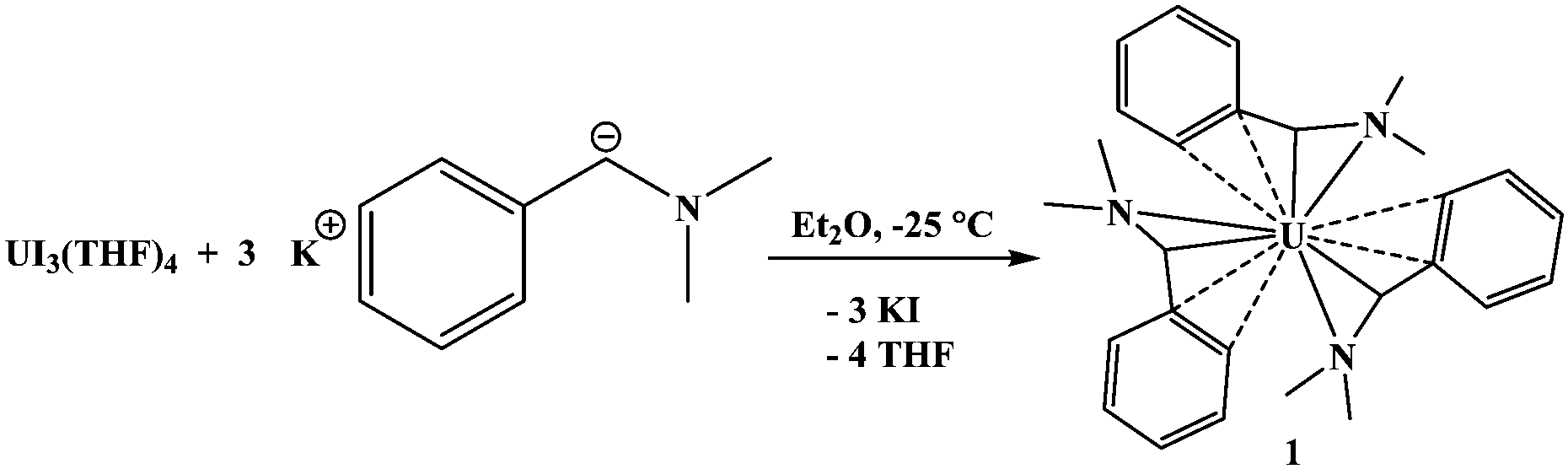 | (1) |
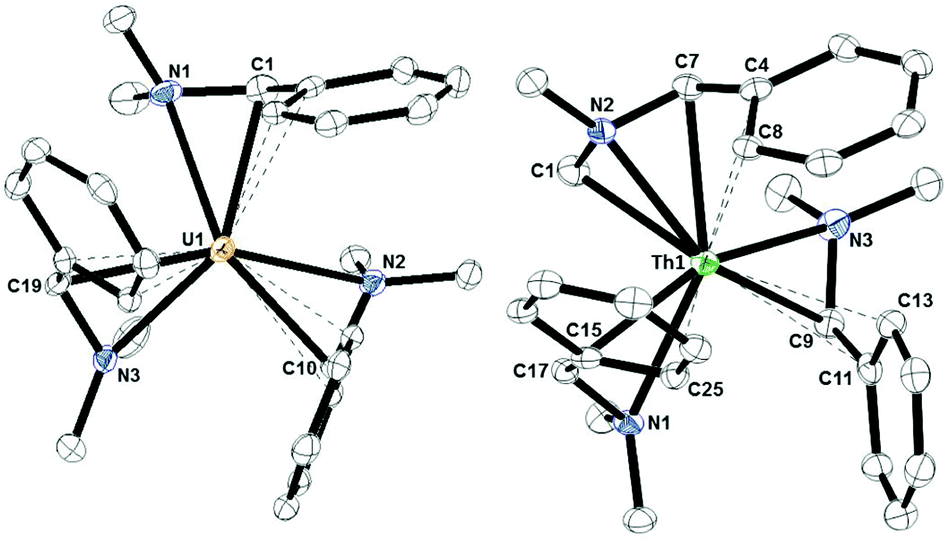 | ||
| Fig. 1 Thermal ellipsoid plot of 1 (left) and 2 (right) shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. | ||
The U–Cipso distances are 2.766(3), 2.784(3), and 2.804(3) Å while the U–Cortho distances are 2.818(3), 2.813(3), and 2.816(3) Å. These distances are far shorter than the closest U–C interactions in U[CH(SiMe3)2]3 but are similar to those found in [U(dddt)3]2−, dddt = 5,6-dihydro-1,4-dithiine-2,3-dithiolate.17 Therefore, the best description of the coordination of the DMBA ligand to uranium is η4-(N,C,C,C) instead of the κ2-(N,C) form resulting from the lithium salt. The uranium-methine carbon bond distances of 2.540(4), 2.521(4), and 2.550(3) Å are between the 2.57(2) Å observed in Tp*2U(CH2C6H5), Tp* = hydrotris(3,5-dimethylpyrazolyl)borate, and 2.48(2) Å in U[CH(SiMe3)2]3.
The magnetization of 1 was studied by variable temperature and variable field experiments. The effective magnetic moment of 1 is shown in Fig. 2. Under Russell–Saunders coupling, U(III) has a 4I9/2 ground multiplet, which is split by the ligand field into substates characterized by mJ. The measured ground state moment of 1 is 1.11 μB, which is in excellent agreement with that of the mJ = 3/2 substate (1.09 μB). Assignment of this ground state to 1 is supported by a failure to observe an EPR spectrum at 2 K as the mJ = 3/2 substate is not EPR active. The first excited state of 1 is ∼100 cm−1 above the ground state as determined from the temperature at which the plot of χT vs. T deviates from linearity. Although U(III) complexes frequently exhibit single molecule magnet (SMM) behaviour,181 does not display a hysteresis in the magnetization vs. field measurements at 2 K. The lack of SSM behaviour is surprising given the ∼100 cm−1 energy of the first excited state. We believe the mechanism for relaxation is tunneling due to dipole–dipole coupling in analogy to the behaviour of U(H2BPz2)3.19 The U–U distance in 1 is 8.0 Å, which is shorter than the 8.2 Å distance in U(H2BPz2)3, which does not display SMM behaviour, and much shorter than that of U(Ph2BPz2)3 (10.8 Å), which does display SMM behaviour.20
 | ||
| Fig. 2 Variable temperature magnetic moment of 1. | ||
Thorium presented an interesting comparison since Th(III) is only accessible using strong reducing agents,21 and since all previously reported compounds of the DMBA ligand needed only three ligands to saturate the coordination sphere. The reaction of ThCl4(DME)2 with four equivalents of K[Me2NC(H)C6H5] at −78 °C, eqn (2), produced an orange solution. The 1H NMR spectrum revealed an asymmetric coordination environment as well as protonated ligand. Orange crystals suitable for X-ray diffraction analysis were obtained from a saturated toluene solution at −25 °C. The structure, Fig. 1, is similar to previous complexes with three DMBA ligands; however, one of the methyl groups has undergone C–H bond activation to afford a dianionic DMBA ligand, Th[η4-Me2NC(H)C6H5]2[η5-(CH2)MeNC(H)C6H5], 2. Similar systems in which U(IV) yields a U(III) product and Th(IV) results in C–H bond activation have been observed previously.22,23
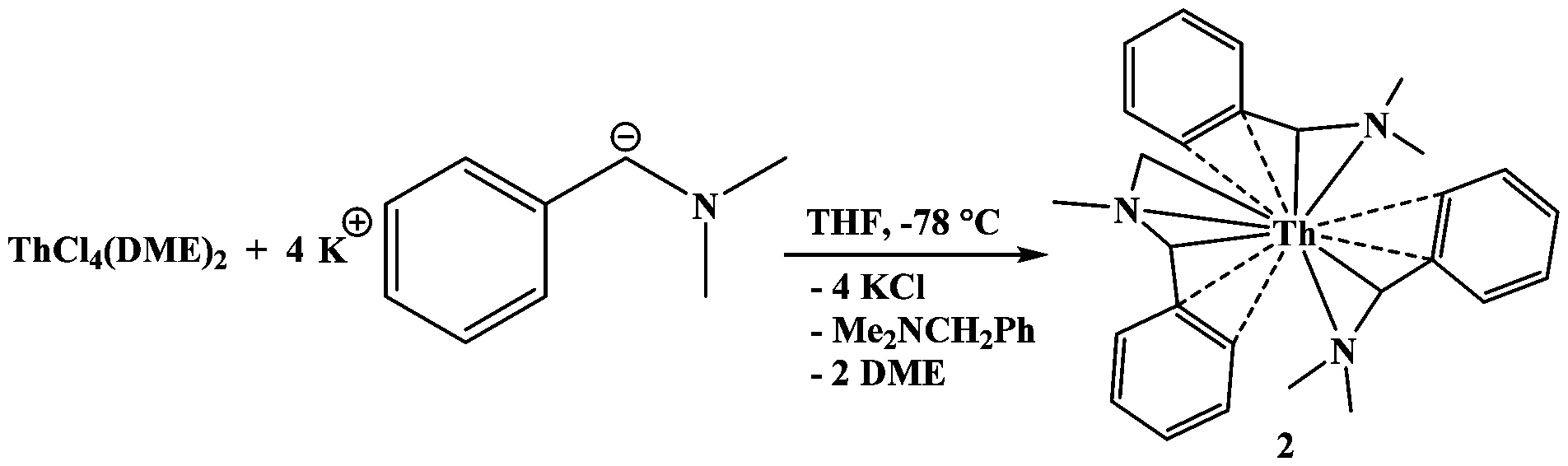 | (2) |
The Th1–C1, Th1–N2, Th1–C7, Th1–C4, and Th1–C8 bond distances are 2.545(4), 2.453(3), 2.578(4), 2.606(3), and 2.866(3) Å, respectively, so the ligand with the C–H bond activated methyl group is a dianionic, η5-ligand. The Th–Cmethine bond distances in 2, 2.578(4), 2.608(4), and 2.620(3) Å, are slightly longer than the Th–Cbenzyl bond length of 2.551(7) Å in (C5Me5)2Th(CH2C6H5)2.24 However, the Th–Cipso bond distances of 2.850(4), 2.908(4), and 2.851(4) Å are significantly shorter than the Th–Cipso bond distance of 2.979(6) Å in (C5Me5)2Th(CH2C6H5)2, but are similar to the 2.700(8)–2.842(4) Å observed for U–Cipso interactions in U(CH2C6H4R)4, R = H, 2-p-iPr; 2-p-tBu; 2-m-OMe; 2-o-picolyl, complexes,6 when the difference in ionic radii are taken into account.
To demonstrate the utility of 1 and 2 as potential starting materials for further substitution, both were treated with three and four equivalents of HS2C[2,6-(Mes)2C6H3], Mes = 2,4,6-Me3C6H2, eqn (3). In both cases, the product is a tetravalent species, An[S2C(2,6-(Mes)2C6H3)]4(THF), An = U, 3; Th, 4, eqn (3). Both 3 and 4 were characterized by X-ray crystallography and were found to be structurally analogous (3 is shown in Fig. 3). Both are nine-coordinate with eight sulfur atoms and one THF molecule completing the coordination sphere in a monocapped square antiprismatic geometry. It is surprising that both thorium(IV) and uranium(IV) are large enough to accommodate four ligands as well as a THF molecule since dithiocarbamate,25 dithiophosphinate,26 and dithiolene17 actinide(IV) complexes are typically eight-coordinate. Our rationale for the presence of the THF molecule is that it may be bound to the metal center prior to or during the addition of the [S2C(2,6-(Mes)2C6H3)]1− ligands, and upon coordination of the dithiocarboxylate ligands, the THF is captured in the inner coordination sphere. The THF molecule cannot be removed by heat or vacuum. The space filling model of the compound is consistent with this explanation as is the observation that both complexes precipitate from the reaction mixture when the reaction is performed in THF. Another interesting feature of complexes 3 and 4 is that typically homoleptic sulfur-based complexes are not produced by protonation reactions. For example, reaction of [{(Me3Si)2N}2U(CH2SiMe2NSiMe3)] with one equivalent of 2,6-Me2C6H3SH yields [(Me3Si)2N]3U[S(2,6-Me2C6H3)], but using four equivalents results in intractable products.27 In our case, reaction of 1 or 2 with four equivalents of HS2C[2,6-(Mes)2C6H3] produced isolable compounds. Both compounds are viable starting materials which may be useful alternatives to the widely used U[N(SiMe3)2]3.28,29
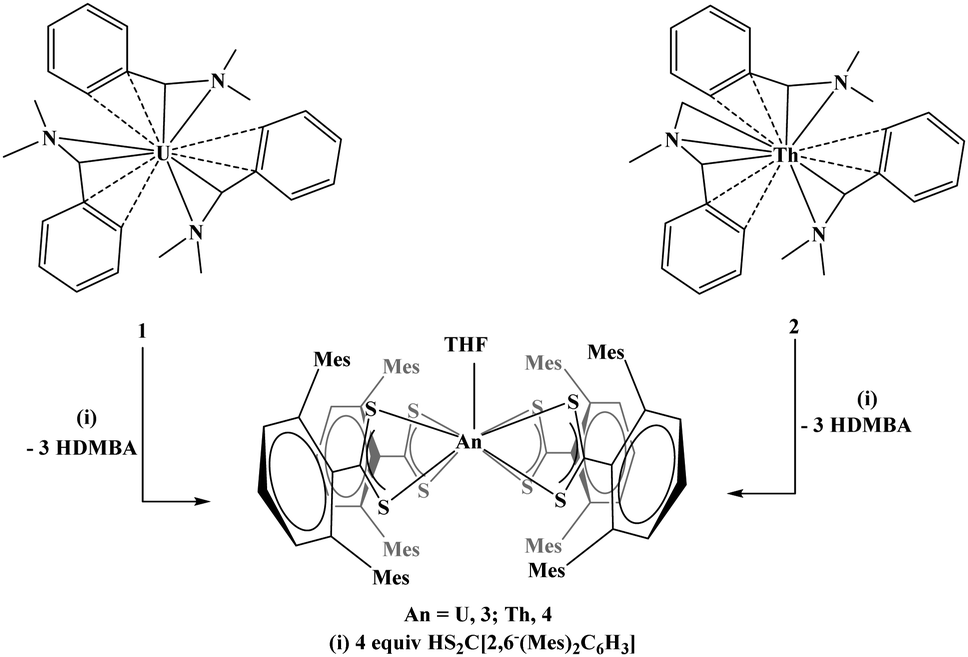 | (3) |
 | ||
| Fig. 3 Thermal ellipsoid plot of 3 shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. | ||
The average U–S bond distances in 3 of 2.8775(17) Å are longer than those seen in [U(dddt)3]2−, which range from 2.717–2.760 Å. This increase is attributed to the greater steric properties of the terphenyl-based ligand. The average Th-S bond distances in 4 of 2.934(3) Å is similar to the 2.932(2) Å distance in the sterically crowded dithiophosphinate complex Th(S2PtBu2)4. These distances are significantly longer than the 2.9075(5) Å and 2.911(4) Å distances in the less crowded complexes Th(S2PiPr2)430 and Th[S2P(C6H11)2]4,31 respectively. The difference in bond distances of 3 and 4 (∼0.057 Å) is consistent with the Shannon radii of nine-coordinate U4+ (1.19 Å) vs. Th4+ (1.23 Å).32
In summary, using the potassium salt of dimethylbenzylamine, we have synthesized and characterized a rare U(III) alkyl complex. When the analogous reaction is attempted with a uranium(IV) starting material, ligand coupling is observed along with reduction to U(III). The thorium complex featured C–H bond activation of one of the methyls on the dimethylamine group. The synthetic utility of these complexes was evaluated using a sterically demanding dithiocarboxylate ligand, HS2C(C6H3Mes2), which produced analogous products, An[S2C(2,6-(Mes)2C6H3)]4(THF), An = U; Th. Further reactivity is currently under investigation.
We gratefully acknowledge the Department of Energy, Office of Science Early Career Research Program under Award DE-SC-0014174 (JRW). WWL was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division (CSGB), Heavy Element Chemistry Program and was performed at Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231.
Notes and references
- L. A. Seaman, J. R. Walensky, G. Wu and T. W. Hayton, Inorg. Chem., 2013, 52, 3556–3564 CrossRef CAS PubMed.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 11732–11743 CrossRef CAS PubMed.
- E. A. Pedrick, P. Hrobarik, L. A. Seaman, G. Wu and T. W. Hayton, Chem. Commun., 2016, 52, 689–692 RSC.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2012, 134, 6160–6168 CrossRef CAS PubMed.
- S. A. Johnson, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Organometallics, 2015, 34, 2889–2895 CrossRef CAS.
- J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS.
- W. G. Van der Sluys, C. J. Burns and A. P. Sattelberger, Organometallics, 1989, 8, 855–857 CrossRef CAS.
- S. Di Bella, G. Lanza, I. L. Fragalà and T. J. Marks, Organometallics, 1996, 15, 205–208 CrossRef CAS.
- E. M. Matson, W. P. Forrest, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2011, 133, 4948–4954 CrossRef CAS PubMed.
- P. G. Edwards, R. A. Andersen and A. Zalkin, Organometallics, 1984, 3, 293–298 CrossRef CAS.
- L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589–10592 CrossRef CAS PubMed.
- E. A. Pedrick, L. A. Seaman, J. C. Scott, L. Griego, G. Wu and T. W. Hayton, Organometallics, 2016, 35, 494–502 CrossRef CAS.
- F. T. Oakes and J. F. Sebastian, J. Organomet. Chem., 1978, 159, 363–371 CrossRef CAS.
- A. C. Behrle and J. A. R. Schmidt, Organometallics, 2011, 30, 3915–3918 CrossRef CAS.
- T. V. Lubben, K. Ploessl, J. R. Norton, M. M. Miller and O. P. Anderson, Organometallics, 1992, 11, 122–127 CrossRef CAS.
- M. Roger, T. Arliguie, P. Thuéry, M. Fourmigué and M. Ephritikhine, Inorg. Chem., 2005, 44, 594–600 CrossRef CAS PubMed.
- F. Moro, D. P. Mills, S. T. Liddle and J. van Slageren, Angew. Chem., Int. Ed., 2013, 52, 3430–3433 CrossRef CAS PubMed.
- K. R. Meihaus, J. D. Rinehart and J. R. Long, Inorg. Chem., 2011, 50, 8484–8489 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS PubMed.
- F. Ortu, A. Formanuik, J. R. Innes and D. P. Mills, Dalton Trans., 2016, 45, 7537–7549 RSC.
- W. J. Evans, J. R. Walensky and J. W. Ziller, Chem. – Eur. J., 2009, 15, 12204–12207 CrossRef CAS PubMed.
- N. A. Siladke, C. L. Webster, J. R. Walensky, M. K. Takase, J. W. Ziller, D. J. Grant, L. Gagliardi and W. J. Evans, Organometallics, 2013, 32, 6522–6531 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682–4692 CrossRef CAS.
- D. Brown, D. G. Holah and C. E. F. Rickard, J. Chem. Soc. A, 1970, 423–425 RSC.
- J. A. Macor, J. L. Brown, J. N. Cross, S. R. Daly, A. J. Gaunt, G. S. Girolami, M. T. Janicke, S. A. Kozimor, M. P. Neu, A. C. Olson, S. D. Reilly and B. L. Scott, Dalton Trans., 2015, 44, 18923–18936 RSC.
- D. L. Clark, M. M. Miller and J. G. Watkin, Inorg. Chem., 1993, 32, 772 CrossRef CAS.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507–1509 CrossRef CAS.
- R. J. Baker, Coord. Chem. Rev., 2012, 256, 2843–2871 CrossRef CAS.
- A. C. Behrle, A. Kerridge and J. R. Walensky, Inorg. Chem., 2015, 54, 11625–11636 CrossRef CAS PubMed.
- A. A. Pinkerton, A. E. Storey and J.-M. Zellweger, J. Chem. Soc., Dalton Trans., 1981, 1475–1480 RSC.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1507960–1507963. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc08105c |
| ‡ Authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2016 |