
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular tweezers target a protein–protein interface and thereby modulate complex formation†
F.
Trusch‡
 a,
K.
Kowski
b,
K.
Bravo-Rodriguez
c,
C.
Beuck
a,
A.
Sowislok
b,
B.
Wettig
b,
A.
Matena
a,
E.
Sanchez-Garcia
c,
H.
Meyer
d,
T.
Schrader
b and
P.
Bayer
*a
a,
K.
Kowski
b,
K.
Bravo-Rodriguez
c,
C.
Beuck
a,
A.
Sowislok
b,
B.
Wettig
b,
A.
Matena
a,
E.
Sanchez-Garcia
c,
H.
Meyer
d,
T.
Schrader
b and
P.
Bayer
*a
aUniversity of Duisburg-Essen, Research Group Structural and Medicinal Biochemistry, Centre for Medical Biotechnology (ZMB), 45117 Essen, Germany. E-mail: peter.bayer@uni-due.de
bUniversity of Duisburg-Essen, Research Group Organic Chemistry, 45117 Essen, Germany
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
dUniversity of Duisburg-Essen, Research Group Molecular Biology I, Centre for Medical Biotechnology (ZMB), 45117 Essen, Germany
First published on 15th November 2016
Abstract
Molecular tweezers for lysine and arginine select a few residues on a protein surface and by their unique complexation mode disrupt a critical protein–protein interaction. Detailed structural information was gained by NMR experiments, strongly supported by QM/MM calculations and further substantiated by ITC, fluorescence anisotropy, ELISA and bio-layer-interference studies.
Modulation of protein–protein interactions (PPI) with small molecules is one of the most promising new approaches in chemical biology and drug discovery.1 To date, most discoveries result from structure-based design, natural product-inspired PPI modulation or compound library generation.2 However, supramolecular ligands are less frequently used for PPI modulation, since the design of compounds which target a predictable well-defined area on a protein is still challenging.3 A promising approach is the construction of compounds which bind single exposed amino acids. In this respect, cyclodextrins, cucurbiturils, resorcinarenes and calixarenes have been used to successfully block critical residues on protein surfaces.4 Recently, we introduced water-soluble supramolecular tweezers highly selective for lysines and arginines (Scheme 1). By a unique threading mechanism, the positively charged side chains of these amino acids are drawn into the tweezers' tailored cavity.5,6 Applications of the tweezers have been so far limited to enzyme inhibition and the disruption of protein–peptide complexes.5,7,8 Here, we report on the inhibition of the protein–protein interaction between the domains of p97 and its cofactor UBXD1 which we use as a model case (for interaction details see SI2, ESI†).
 | ||
| Scheme 1 Structure of the molecular tweezers with included lysine side chain inside the cavity. | ||
Specifically, we reveal the affinity profile of the tweezers to preferred lysine and arginine residues on the p97-N surface with NMR spectroscopy in combination with Molecular Dynamics (MD) and Quantum Mechanics/Molecular Mechanics (QM/MM) calculations. Subsequent biophysical assays show how binding of the tweezers to p97-N disrupts the interaction with UBXD1-N.
p97 (also called VCP) is an abundant AAA-ATPase involved in cell cycle regulation, DNA repair and protein degradation.9 Accordingly, missense mutations in p97 cause degenerative diseases.10 Its N-terminal domain p97-N1–199 with 32 basic residues represents an attractive target for the molecular tweezers (Fig. S1, ESI†). Hence, we performed 2D NMR experiments to elucidate whether binding to the mainly positively charged protein surface is unspecific or if the tweezers occupy preferred well-defined interaction sites. Sub-stoichiometric addition of tweezers to 15N-labeled p97-N up to a molar ratio of 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 produced shifting as well as disappearing NH-signals in a series of 1H–15N-HSQC spectra (Fig. S2, ESI†). Especially decreasing signal intensities indicate specific interactions,8 while chemical shift changes may also occur as averaged values when ligands explore the protein surface mainly guided by electrostatic interactions in a process termed “protein surface camouflage”.11 When mapping the corresponding amino acids on the surface of the p97-N structure, six well-defined areas (patches 1–6) were identified; likely accommodating one tweezers molecule each (Fig. 1a and Table 1).
:1 produced shifting as well as disappearing NH-signals in a series of 1H–15N-HSQC spectra (Fig. S2, ESI†). Especially decreasing signal intensities indicate specific interactions,8 while chemical shift changes may also occur as averaged values when ligands explore the protein surface mainly guided by electrostatic interactions in a process termed “protein surface camouflage”.11 When mapping the corresponding amino acids on the surface of the p97-N structure, six well-defined areas (patches 1–6) were identified; likely accommodating one tweezers molecule each (Fig. 1a and Table 1).
 | ||
| Fig. 1 Tweezers interaction sites with p97-N. (a) According to 1H–15N-HSQC experiments, tweezers interact specifically with lysines and arginines on the p97-N surface (PDB: 1E32, full length p97). Residues of p97-N (260 μM) that interact with the tweezers (130 μM) are restricted to 6 patches (rose: interacting residues of p97-N; white: non-interacting residues of p97-N; grey: non-interacting residues of the ND1-linker). Patches are indicated (p1–p6, white) with the tweezers hosting residue of each patch (black). (b) QM/MM calculations reveal a very stable inclusion complex between K20 (flexible N-terminus of p97-N) and the tweezers. (c) QM/MM calculations of the tweezers around the loop-exposed K63 also indicate the formation of a very stable complex. Note the additional favourable interactions between the tweezers' hydrogen phosphate groups and neighbouring arginine residues around K20 and K63. | ||
| Patch | Binding sitea | Host residueb | ASc | K D [μM] | 〈KD〉d [μM] |
|---|---|---|---|---|---|
| a Binding site: residues in a patch whose NH-signals undergo chemical shift or intensity changes upon titration with tweezers. Corresponding areas (p1–p6) are shown in Fig. 1a. b Host residue: highlights the residue which is most likely bound by the tweezers within a patch, explaining all peak perturbations in 1H–15N-HSQC titration experiments. *Host residue discriminated by QM/MM calculations. c AS: selected residues from shift perturbation analysis with NH-titration curves suitable for non-linear regression to estimate dissociation constants (KD). d 〈KD〉: averaged KD or weighted KD ranges for the whole patch bound by one tweezers ligand. | |||||
| 1 | I16, L17, K18, Q19, K20, R22, R25, G97, V99, I100, S101, I102, Q103 | K20* |
V99
I100 S101 |
60
70 50 |
60 |
| 2 | K60, G61, K63, R93 |
K63*/
K60 |
K60 | 110 | 70–80 |
| G61 | 20 | ||||
| R93 | 100 | ||||
| 3 | R113, I114, H115, L117, K148, V165, T168 |
K148*/
R113 |
I114 | 10 | 30–80 |
| L117 | 60 | ||||
| V165 | 30 | ||||
| R113 | 100 | ||||
| 4 | A31, E34, D35, S37, R86 |
R86* (R144/
R155) |
A31 | 100 | 80–90 |
| D35 | 120 | ||||
| S37 | 30 | ||||
| 5 | I119, D120, D121, V123, R159, E162, K164 |
R159*/
K164 |
D120
V123 |
10
75 |
30–40 |
| 6 | K190, R191, E195, E196 | K190 | K190 | 40 | 50 |
| R191 | 80 | ||||
| E196 | 30 | ||||
Due to the extensive aromatic system of the tweezers, several NH-signals of residues around a bound lysine or arginine also show changes. Analysis and subsequent mapping of resonances are in some cases ambiguous since some patches contain more than one positively charged amino acid. With NMR data we could assign host residues for tweezers binding as K20, K60/K63, R113/K148, R86/R144/R155, R159/K164 and K190. Chemical shifts and intensity changes of NH-signals of all involved amino acids within the preferred binding regions produced titration curves suitable for nonlinear regression with dissociation constants in the lower to medium μM-range (Table 1 and Fig. 1a).
To retrieve thermodynamic parameters, binding of tweezers to p97-N was also monitored by isothermal titration calorimetry (ITC) (Fig. 2).
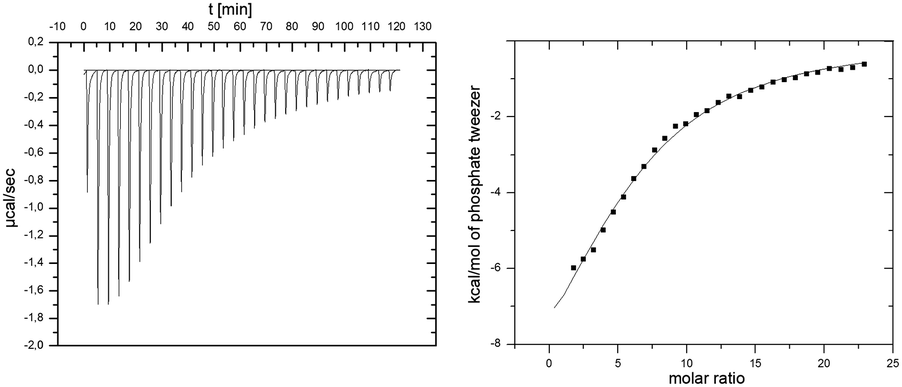 | ||
| Fig. 2 ITC experiment of p97-N and added tweezers. 1 mM tweezers were titrated against 10 μM p97-N. Data was fitted with a multiple-site binding model without a given N. | ||
The binding process is enthalpically driven as indicated by a strong negative ΔH = −64.1 kcal mol−1 that overcompensates the unfavorable entropic changes of −TΔS = 56.9 kcal mol−1. This phenomenon is typical for the specific inclusion inside the tweezers' cavity at the cost of reduced mobility of the amino acid side chain. The analysis of calorimetric data regarding the molar ratio between tweezers and p97-N results in a stoichiometry of 6 tweezers per p97-N molecule, which is in remarkable agreement with the above discussed 1H–15N-HSQC-titration experiment (Fig. 1a). From the averaged Gibbs free energy of −7.2 kcal mol−1 for each single binding event, the average KD value was calculated to 6 μM, which is slightly lower than the estimated affinities from NMR titration experiments (Table 1).
To rule out the remaining ambiguities between two potential host residues in the same patch and to reappraise the tweezers binding sites on p97-N, MD simulations were performed followed by QM/MM calculations. Of the 26 lysines and arginines included in the crystal structure (PDB: 3QQ7), only 13 formed conserved inclusion complexes during MD simulations. In all these cases, the host residue side chain was well threaded into the cavity of the tweezers (Fig. S3 and Table S1, ESI†). For these residues, QM/MM calculations were performed (for details see SI1, ESI†) whereby the electronic energies of the QM regions reflect the relative stability of the tweezers–amino acid complexes depending of the molecular environment. The subsequent analysis allowed assigning of the preferred tweezers binding residue in each patch (Table S2, ESI†). In patch 1, K20 is determined as the host residue by NMR experiments and has also one of the lowest QM energies (Fig. 1b and Table S2, ESI†). The QM/MM calculations also qualify K63 over K60 as the favourite tweezers binding site in patch 2. Unlike in K60, the side chain of K63 is solvent exposed and well accessible. In addition, positively charged neighbouring amino acids are able to interact with the negatively charged tweezers' free hydrogen phosphate group (Fig. 1c). NMR data point towards K148 or R113 as a third putative tweezers host residue (patch 3). Although both complexes are not conserved during the MD simulations, the positive electrostatic surface potential of p97-N formed by several K148-surrounding arginines and lysines (R64, R65, R147, K164; Fig. S1d, ESI†) contributes to keep the tweezers close to the protein surface. This most likely causes the strong non-inclusion interaction around K148 seen by NMR experiments (Fig. S1c, ESI†). QM/MM calculations predict R86 of patch 4 next to the primary protein interaction site of p97-N, the VIM groove, to form one of the most stable of all arginine–tweezers complexes (Table S2, ESI†). This arginine is well solvent-exposed without steric impediment and is supported by secondary electrostatic interactions between the tweezers' hydrogen phosphate groups and positively charged residues in close proximity. For patch 5, QM/MM calculations clearly favour R159 over K164, because it forms a conserved inclusion complex with the tweezers, despite the lack of secondary interactions with surrounding residues. Taken together, we propose K20, K63, K148, R86, R159 and K190 as the preferred targets of the tweezers on p97-N (Table 1, patches 1–6) with binding to K148 taking place via non-inclusion electrostatic interactions. To rule out steric hindrance of the tweezers while simultaneous binding, we performed MD simulations with preformed inclusion complexes of the tweezers on K20, K63, R86 and R159 and a non-inclusion complex with K148 (K190 is not included in the crystal structure of p97-N used for the simulations) (Fig. 3 and Fig. S4, ESI†). All inclusion complexes were conserved and the tweezers established secondary interactions as described for the 1:1 complexes. These results confirm that there is sufficient space at the surface of p97-N to host five tweezers simultaneously and, most likely, a sixth one in the flexible C-terminus around K190.
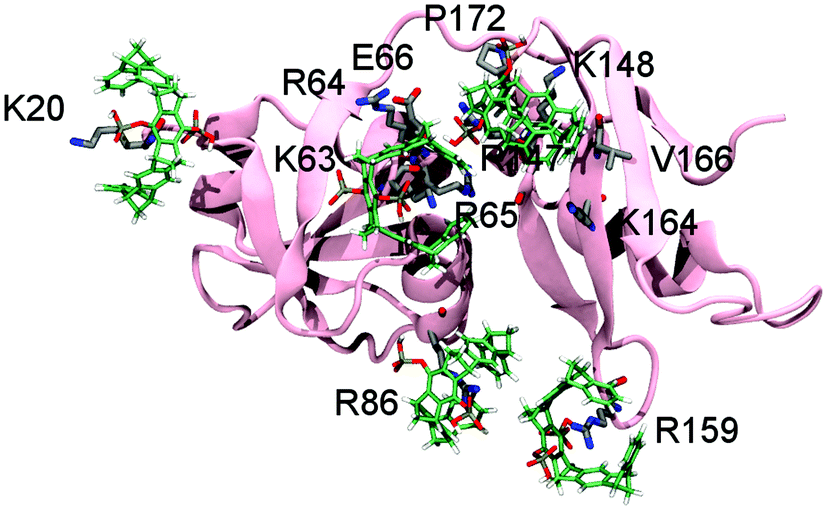 | ||
| Fig. 3 Simultaneous binding of tweezers on p97-N. There is enough space at the surface of p97-N to allow the simultaneous binding of several tweezers in the positions predicted by QM/MM calculations (the last frame from one of the MD simulations is shown). The inclusion complexes with K20, K63, R86 and R159 were conserved and the fifth tweezer molecule remains around K148, as in the 1:1 complex. | ||
Since residues of both the VIM groove and the ND1-linker of p97-N are targeted by the tweezers (Fig. 1a and SI2, ESI†), but also mediate adaptor binding of UBXD1-N,12 we investigated the potential disrupting effect of the tweezers on the complex formation between p97-N and UBXD1-N by fluorescence anisotropy, ELISA and BLI (Fig. 4). First, complex formation of p97-N with labelled UBXD1-N was monitored by fluorescence anisotropy changes in the presence of up to 50 mol% tweezers. Protein and tweezers ratios were carefully optimized to avoid the formation of protein aggregates. The apparent KD value ascends from 9 μM (no tweezers) to 216 μM already at low tweezers concentrations (Fig. 4a and Fig. S5, ESI†). Importantly, conformational changes on p97-N during tweezers binding as an alternative explanation for the weakened protein–protein interaction with UBXD1-N were excluded by CD spectroscopy (Fig. S7, ESI†). The inhibiting character of the tweezers on the UBXD1-N/p97-N complex formation was also confirmed by ELISA experiments. Immobilized UBXD1-N (30–210 nM) was incubated with combinations of p97-N and varying tweezers concentrations (Fig. 4b). Increasing tweezers concentrations substantially impeded complex formation between UBXD1-N and p97-N.
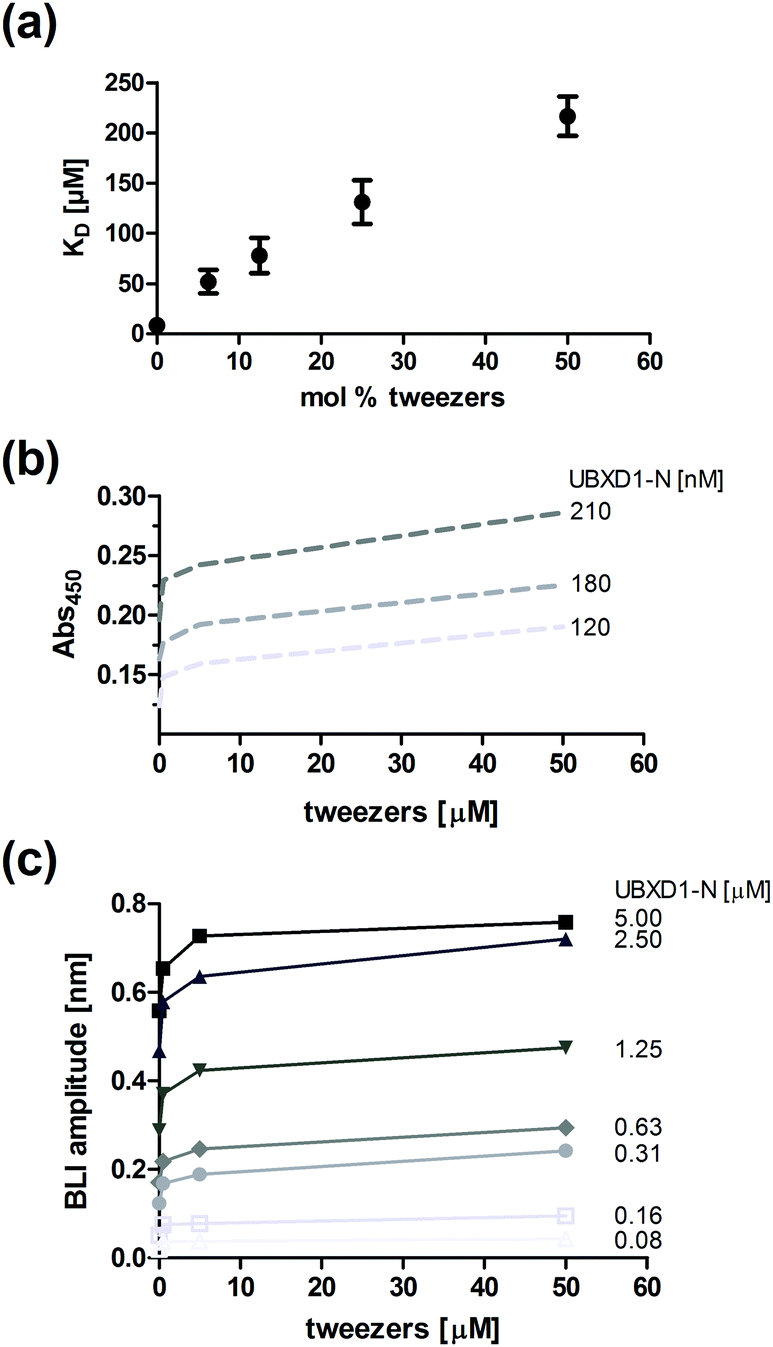 | ||
| Fig. 4 Complex modulation of p97-N and UBXD1-N in the presence of the tweezers. (a) Affinity determination between UBXD1-N and p97-N by fluorescence anisotropy after pre-incubation of p97-N with increasing amounts of tweezers. (b) ELISA experiment with immobilized UBXD1-N (120, 180 and 210 nM exemplarily shown) and a constant p97-N concentration (1.2 μM) in the presence of varying tweezers concentrations (0–50 μM). (c) Steady-state analysis of bio-layer-interference (BLI) experiments titrating UBXD1-N as indicated to immobilized p97-N in the presence of different tweezers concentrations (0–50 μM). | ||
In bio-layer interference (BLI) experiments, titrations of immobilized p97-N with increasing concentrations of UBXD1-N (0–5 μM) were performed in the presence of different tweezers concentrations. The apparent KD value obtained by steady-state data analysis increased from 2.1 μM to 21 μM (Fig. 4c and Fig. S6, ESI†). The effective concentration of the tweezers in ELISA and BLI experiments is likely overestimated because a fraction is also bound by the blocking agents. Nevertheless, the system reaches saturation at large tweezers excess, indicating specific binding to the protein–protein interface (Fig. 4b and c). In total, the three different experimental approaches confirm that the formation of tweezers–lysine/arginine inclusion complexes can significantly impair the interaction between two protein domains.
We also tested the ability of our supramolecular ligand to target the binding partner UBXD1-N by titrating 15N-labeled UBXD1-N with increasing amounts of tweezers (Fig. S8, ESI†). Since this moiety of UBXD1 is disordered, most lysine residues are bound by the tweezers. Although the critical VIM motif in UBXD1 which binds to the VIM groove of p97-N (SI2, ESI†) does not contain any lysine or arginine residues, especially the adjacent K66 may also contribute to weaken the important p97-N/UBXD1-N interaction.
In summary, hydrogen phosphate tweezers are binding at sub-stoichiometric concentrations with low micromolar KD values preferably to basic lysines and arginines restricted to 6 patches on the p97-N protein surface. The tweezers binding is driven most likely by a combination of electrostatic interactions, hydrogen bonds, dispersive attraction and hydrophobic effects.13 Furthermore, QM/MM calculations confirmed preferred complexation sites but also allowed to discriminate between ambiguous host residues derived from NMR data. The inhibition of the protein–protein interaction between UBXD1-N and p97-N by the tweezers was confirmed by fluorescence anisotropy. ELISA and BLI experiments further support the weakening of the protein–protein interaction. In contrast to other supramolecular ligands like calixarenes and PTS (pyrene-tetrasulfonic acid)14 the tweezers do not promote unspecific formation of protein assemblies. Thus, they may be amenable for specific inhibition of selected binding sites upon further chemical variation.
Notes and references
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695 CrossRef CAS PubMed.
- R. Kubotaa and I. Hamachi, Chem. Soc. Rev., 2015, 44, 4454 RSC.
- D. A. Uhlenheuer, K. Petkau and L. Brunsveld, Chem. Soc. Rev., 2010, 39, 2817 RSC.
- F.-G. Klärner, B. Kahlert, A. Nellesen, J. Zienau, C. Ochsenfeld and T. Schrader, J. Am. Chem. Soc., 2006, 128, 4831 CrossRef PubMed.
- M. Fokkens, T. Schrader and F.-G. Klärner, J. Am. Chem. Soc., 2005, 127, 14415 CrossRef CAS PubMed.
- F.-G. Klärner, T. Schrader, J. Polkowska, F. Bastkowski, P. Talbiersky, M. Campañá Kuchenbrandt, T. Schaller, H. de Groot and M. Kirsch, Pure Appl. Chem., 2010, 82, 991 CrossRef.
- D. Bier, et al. , Nat. Chem., 2013, 5, 234 CrossRef CAS PubMed.
- D. S. Haines, Genes Cancer, 2010, 1, 753 CrossRef CAS PubMed; M. Livingstone, et al. , Cancer Res., 2005, 65, 7533 Search PubMed; H. Meyer, M. Bug and S. Bremer, Nat. Cell Biol., 2012, 14, 117 CrossRef PubMed.
- H. Meyer and C. C. Weihl, J. Cell Sci., 2014, 127, 3877 CrossRef CAS PubMed.
- R. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527 CrossRef CAS PubMed; R. E. McGovern, B. D. Snarr, J. A. Lyons, J. McFarlane, A. L. Whiting, I. Paci, F. Hof and P. B. Crowley, Chem. Sci., 2015, 6, 442 RSC.
- F. Trusch, A. Matena, M. Vuk, L. Koerver, H. Knævelsrud, P. S. Freemont, H. Meyer and P. Bayer, J. Biol. Chem., 2015, 290, 29414 CrossRef CAS PubMed.
- F.-G. Klärner and T. Schrader, Acc. Chem. Res., 2013, 46, 967 CrossRef PubMed.
- H. P. Morgan, I. W. McNae, K. Y. Hsin, P. A. Michels, L. A. Fothergill-Gilmore and M. D. Walkinshaw, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 215 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc08039a |
| ‡ Present address: University of Aberdeen, Institute of Medical Sciences, Foresterhill, AB25 2ZD, Aberdeen, UK. |
| This journal is © The Royal Society of Chemistry 2016 |