
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Duplications of an iron–sulphur tripeptide leads to the formation of a protoferredoxin†
Simone
Scintilla
a,
Claudia
Bonfio
a,
Luca
Belmonte
a,
Michele
Forlin
a,
Daniele
Rossetto
a,
Jingwei
Li
b,
James A.
Cowan
b,
Angela
Galliani
c,
Fabio
Arnesano
c,
Michael
Assfalg
d and
Sheref S.
Mansy
 *a
*a
aCIBIO, University of Trento, Via Sommarive 9, 38123 Povo, Italy. E-mail: mansy@science.unitn.it
bDepartment of Chemistry and Biochemistry, The Ohio State University, 100 West 18th Ave, Columbus, OH 43210, USA
cDepartment of Chemistry, University of “Bari A. Moro”, Via E. Orabona 4, 70125 Bari, Italy
dDepartment of Biotechnology, University of Verona, Strada Le Grazie 15, 37134 Verona, Italy
First published on 25th October 2016
Abstract
Based on UV-Vis, NMR, and EPR spectroscopies and DFT and molecular dynamics calculations, a model prebiotic [2Fe–2S] tripeptide was shown to accept and donate electrons. Duplications of the tripeptide sequence led to a protoferredoxin with increased stability. Duplications of primitive peptides may have contributed to the formation of contemporary ferredoxins.
Iron–sulphur clusters are thought to be among the most ancient of biological cofactors. Iron and sulphide were abundant on prebiotic Earth, and iron and sulphide readily assemble into iron–sulphur clusters in the presence of thiolate ligands.1 The protein conformation most commonly associated with iron–sulphur cluster coordination, i.e. the βαββαβ ferredoxin fold, is widespread in biology, suggesting that the fold itself is ancient.2–5 [2Fe–2S] and [4Fe–4S] ferredoxins play important roles in electron transfer reactions and participate in central metabolic pathways including those that lead to the formation of a proton gradient that is used, in part, to drive the synthesis of ATP.6
Before the existence of complex protein folds, life must have relied on simpler, readily available catalysts. It has been proposed that iron–sulphur containing minerals could have played such a role and thus helped shape protometabolic processes that were later mediated by iron–sulphur proteins.6–9 However, the transition from mineral to biological cofactor is unclear. One possibility is that small, redox active iron–sulphur peptides grew into longer chains with increased stability and activity through duplication events. Eck and Dayhoff suggested that the modern day ferredoxin evolved through such iterative duplications starting from a tetrapeptide sequence.10 More recent computational studies have proposed that a conserved CxxCxxC motif involved in the coordination of a [4Fe–4S] cluster may have existed within a putative protoferredoxin sequence.11,12 These proposals seem plausible, because short peptides can coordinate iron–sulphur clusters. For example, amino- and carboxy-terminally blocked CxxC peptides bind [2Fe–2S] clusters in dimethyl sulfoxide, and the tripeptide glutathione (γECG) stabilizes the formation of a [2Fe–2S] cluster in aqueous solution.13,14 In each of these cases, multiple peptides coordinate to the same [2Fe–2S] in order to provide the four thiolate ligands needed to stabilize the cluster.
To test whether a protoferredoxin-like polymer could emerge from a short peptide, we characterized [2Fe–2S] glutathione and longer [2Fe–2S] peptides composed of repeating glutathione units (Fig. 1). Glutathione was used as a model prebiotic tripeptide, because this peptide has already been shown to coordinate an iron–sulphur cluster and because glutathione is readily available. Our data show that [2Fe–2S] tripeptide complexes are highly dynamic, exhibit redox activity, and become more stable upon polymerization. Further, polymers composed of repeating tripeptide units give cysteinyl ligand spacing similar to contemporary ferredoxins.
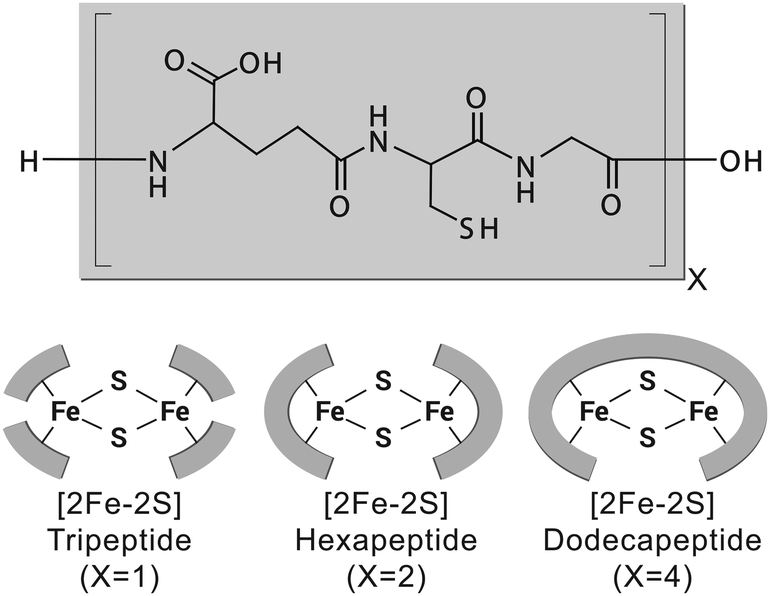 | ||
| Fig. 1 Glutathione (top, grey block) can stabilize the formation of a [2Fe–2S] cluster (X = 1, bottom left). Two and four duplications of the tripeptide lead to longer [2Fe–2S] polymers (X = 2 and 4, bottom centre and bottom right, respectively). | ||
The optimal stoichiometry of glutathione, FeCl3, and Na2S was determined for cluster assembly on the tripeptide glutathione. Titrations were monitored by the decomposition of UV-visible absorption spectra (Fig. S1, ESI†). A stoichiometric amount of each component did not give a [2Fe–2S] under anaerobic conditions. 1H NMR spectroscopy showed that instead of cluster formation, glutathione became oxidized, presumably by donating electrons to the ferric ions (Fig. S2, ESI†). At an iron ion concentration of 0.5 mM, a maximum amount of [2Fe–2S] was formed with 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.4 glutathione:FeCl3:Na2S (Fig. S3–S5, ESI†). The requirement for excess glutathione was confirmed by chromatography with a glutathione conjugated sepharose resin. The eluate containing [2Fe–2S] glutathione was detected only when free glutathione was present in the running buffer (Fig. S6, ESI†). [2Fe–2S] glutathione showed a UV-visible absorption spectrum similar to that of human ferredoxin (Fig. 2), and the mass spectrometry was consistent with four glutathiones coordinated to one [2Fe–2S] (Fig. S7, ESI†), as previously reported.15 A pH titration revealed that [2Fe–2S] glutathione persisted between pH 7.5 and pH 10, with a maximum at pH 8.5 (Fig. S8, ESI†). The cluster was stable to 0.5 M NaCl and MgCl2 (Fig. S9, ESI†).16
:1:0.4 glutathione:FeCl3:Na2S (Fig. S3–S5, ESI†). The requirement for excess glutathione was confirmed by chromatography with a glutathione conjugated sepharose resin. The eluate containing [2Fe–2S] glutathione was detected only when free glutathione was present in the running buffer (Fig. S6, ESI†). [2Fe–2S] glutathione showed a UV-visible absorption spectrum similar to that of human ferredoxin (Fig. 2), and the mass spectrometry was consistent with four glutathiones coordinated to one [2Fe–2S] (Fig. S7, ESI†), as previously reported.15 A pH titration revealed that [2Fe–2S] glutathione persisted between pH 7.5 and pH 10, with a maximum at pH 8.5 (Fig. S8, ESI†). The cluster was stable to 0.5 M NaCl and MgCl2 (Fig. S9, ESI†).16
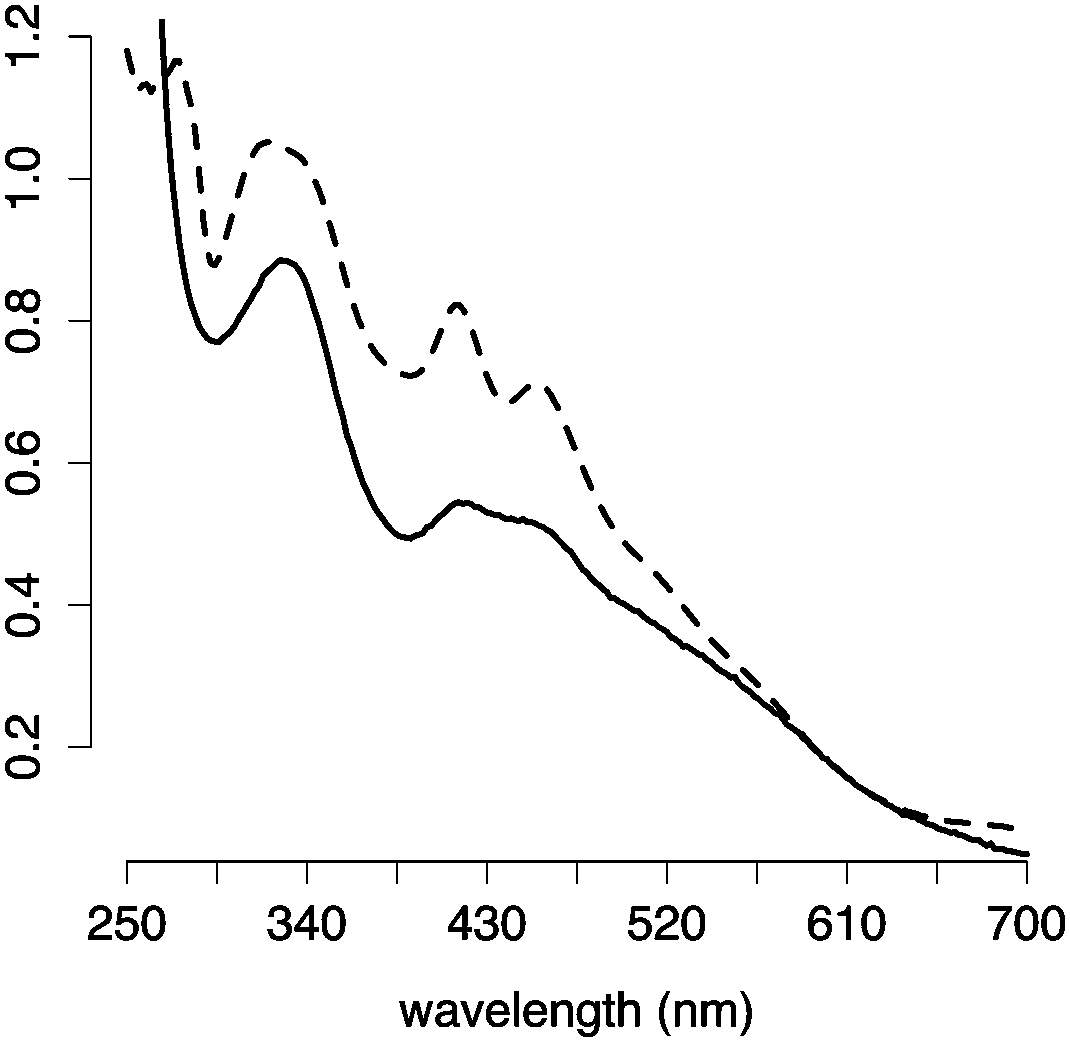 | ||
| Fig. 2 UV-visible absorption spectra of [2Fe–2S] glutathione (solid line) and human ferredoxin (dashed line). | ||
The large excess of peptide that was needed to stabilize the iron–sulphur cluster suggested that the rate of ligand exchange was high. To gain more insight into the dynamics of the system, [2Fe–2S] glutathione was assessed by NMR spectroscopy. Even though the ratio of free glutathione to cluster coordinated glutathione was 80:1, only one set of proton resonance peaks was observed, consistent with ligand exchange within hundreds of milliseconds. The main difference in the diamagnetic region was the broadening of the peaks with respect to glutathione in the absence of the [2Fe–2S] (Fig. S10, ESI†). The extent of broadening and the uneven quenching of the resonance amplitudes was consistent with interactions with paramagnetic centres. In fact, we were able to observe the paramagnetic shifted resonances of the α and β protons of cysteine at 11 ppm and 30–40 ppm (Fig. 3a) by decreasing the recycle delay and the acquisition time of the experiments. The detected hyperfine resonances were similar to those previously observed for reduced, i.e. [2Fe–2S]1+, ferredoxin.17
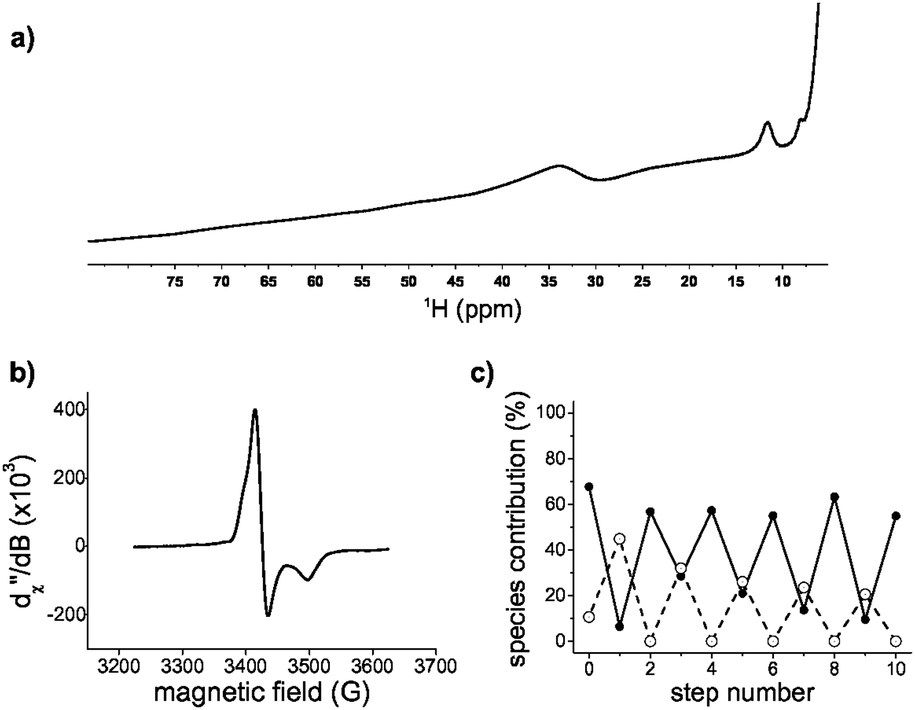 | ||
| Fig. 3 [2Fe–2S] glutathione is redox active. (a) 1H-NMR paramagnetic spectrum of [2Fe–2S]1+ glutathione. (b) EPR spectrum of [2Fe–2S]1+ glutathione at 40 K. The values of g 1.96, 2.00, and 2.02 are consistent with a mainly axial system with small rhombicity. (c) [2Fe–2S]2+ (filled circles) and [2Fe–2S]1+ (open circles) glutathione during reduction and oxidation determined by UV spectral decomposition. | ||
Since the NMR data suggested the presence of a reduced cluster, whereas previous Mössbauer measurements on freshly prepared sample were consistent with an oxidized cluster,14 we probed whether the [2Fe–2S] coordinated to glutathione was redox active. Oxidized clusters, i.e. [2Fe–2S]2+, are EPR silent because of antiferromagnetic coupling, whereas reduced ferredoxin-like centres show a distinctive g = 1.9 signal.18 The addition of the reductant dithionite to a freshly prepared sample showed EPR features consistent with a [2Fe–2S]1+ coordinated to glutathione (Fig. 3b and Fig. S11 and S12, ESI†). To determine whether the reduced state was stable and capable of returning to the oxidized state, a reduced sample was run through a Sephadex G-10 gel filtration column to remove the reductant and any degraded cluster that may have formed. The eluate was then oxidized with hydrogen peroxide. The resulting UV-visible absorption spectrum showed the presence of [2Fe–2S]2+ glutathione, confirming that [2Fe–2S] glutathione can go through one complete round of reduction-oxidation (Fig. S11, ESI†). Several rounds of reduction and oxidation with dithionite and hydrogen peroxide were possible with minimal degradation of the iron–sulphur cluster (Fig. 3c). The data were consistent with the ability of a tripeptide to mimic the cluster coordination and redox activity of ferredoxin proteins.
Since a tripeptide displayed some of the properties of a ferredoxin, we wondered if longer polymers consisting of repeating tripeptide units would lead to a more stable, protoferredoxin-like sequence. However, in order for such a mechanism to work, the spatial orientation of the peptidyl ligands needs to be preserved from the transition of a tripeptide to longer polymers. Extracting such detailed structural information from experimental data was not practical because of the dynamics of the complex and the paramagnetism of the cluster. Therefore, a computational model was built. Density functional theory (DFT) calculations were used to map the parameters of the [2Fe–2S] by using coordinates from the deposited crystal structure of glutaredoxin as a starting point. Glutaredoxin contains a [2Fe–2S] coordinated to two cysteine ligands provided by two glutathione molecules and two cysteines from protein subunits. To simplify the calculations, each ligating cysteine was truncated at the β carbon, as previously described.19 Calculations with GAMESS-US20,21 gave a [2Fe–2S] structure that superimposed with the [2Fe–2S] of glutaredoxin with a RMSD of 1.0 Å. The calculated CHARMM parameters were then used for molecular dynamics simulations with NAMD to probe the conformation of the complex composed of four glutathiones and one [2Fe–2S]. An alignment of the entire simulation trajectory showed a low RMSD (3.9 ± 0.4 Å), consistent with the presence of a geometrically stable structure. The four individual glutathiones coordinated to the [2Fe–2S] showed similar folds (RMSDs between 0.53 Å and 0.60 Å).
The structure of [2Fe–2S] glutathione suggested that polymerization into longer peptide polymers would not interfere with the geometric stability of the molecule. The three-dimensional conformation of the complex as a whole was not globular, exhibiting a largely planar shape. The structure was stabilized by intramolecular backbone electrostatic contacts including strong hydrogen bonding between cysteine and glutamate that persisted for the entire trajectory. There were also a series of intermolecular interactions that persisted in 17% to 50% of the trajectory. One such interaction was between the backbone amino group of glutamate and the backbone carboxylate of glycine of an adjacent glutathione, as previously predicted (Fig. 4).15,16 These two functional groups were less than 3.9 Å apart in 10% of the trajectory (Table S2, ESI†). Such spacing suggested that the incorporation of a peptide bond at this position would lead to longer polymers without significant disruption to the overall structure of the complex. To gain more insight into this possibility, repeating units of the tripeptide, including a hexapeptide and a dodecapeptide, were modelled. The RMSD of two hexapeptides and one dodecapeptide superimposed with the structure of four glutathiones coordinated to a [2Fe–2S] was 3.9 Å and 3.4 Å, respectively. If only the cysteine side-chain (Cβ and Sγ) ligands and [2Fe–2S] were taken into consideration, the RMSD was 0.9 Å for the hexamer and 0.8 Å for the dodecamer.
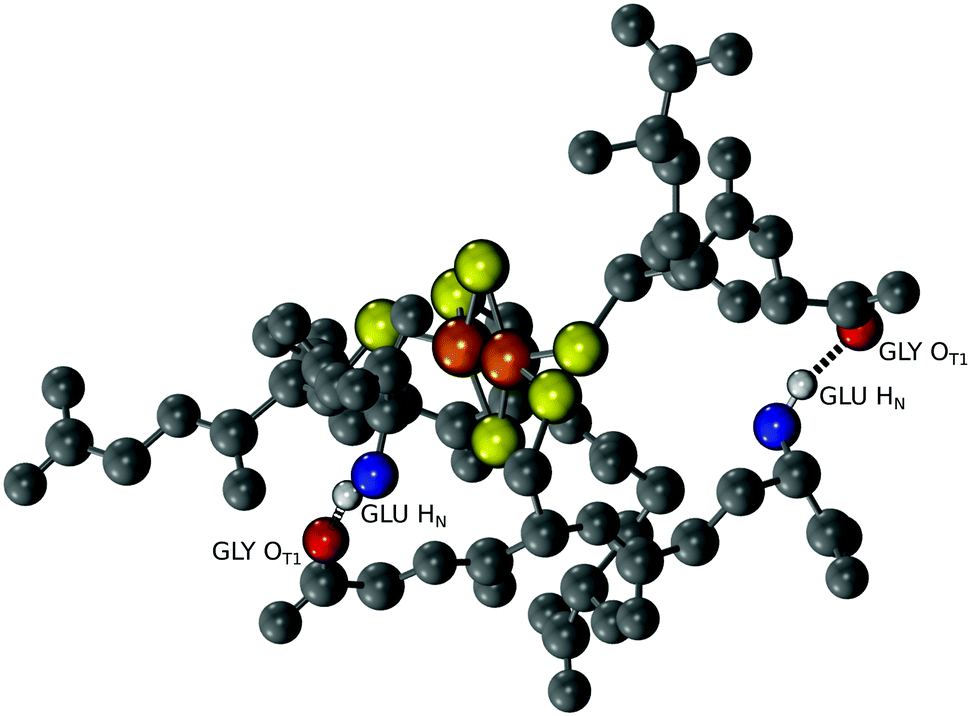 | ||
| Fig. 4 Molecular dynamics show interactions between the amino group of glutamate and the carboxy group of glycine of adjacent glutathione molecules coordinated to the same [2Fe–2S]. A snapshot of the trajectory is shown in which the hydrogen bonds are depicted as black springs. | ||
Since the modelling data suggested that repeating units of glutathione did not disrupt the structure of the complex, we probed whether duplication events could lead to [2Fe–2S] peptides of greater stability. Peptides of six, nine, and twelve amino acids in length corresponding to polymers consisting of two, three, and four glutathione units were synthesized (Fig. S13–S15 and Table S3, ESI†) and evaluated for [2Fe–2S] binding and stability. All three peptides were capable of coordinating a [2Fe–2S], although the [2Fe–2S] coordinated to the nonapeptide rapidly degraded (Fig. S16–S18, ESI†). This was consistent with the structural model. The stoichiometry between ligands and the [2Fe–2S] was fully satisfied by either two hexapeptides or one dodecapeptide in a manner that did not disrupt the overall structure of the complex. Conversely, a single nonapeptide would not provide enough cysteine ligands for cluster coordination, and two nonapeptides would distort the structure of the complex. Nevertheless, duplication events leading to a protoferredoxin would not have required passage through a nonapeptide state. A duplication of a tripeptide followed by a duplication of the resulting hexapeptide would give the dodecapeptide without an intermediate, less stable nonapeptide transition.
Polypeptides containing repeating sequences of glutathione are more stable and contain ferredoxin-like cysteine spacing. [2Fe–2S] peptide stability was assessed in two ways, by determining the amount of excess peptide required to stabilize the cluster and by measuring the kinetics of the degradation of the [2Fe–2S]. For solutions containing 0.25 mM iron and 0.1 mM sulphide, the minimum amount of ligand needed to form the [2Fe–2S] was 20 mM for glutathione and 1 mM for both the hexamer and the dodecapeptide (Fig. 5a and Fig. S19, ESI†). Comparison of the t1/2 of the [2Fe–2S] for each peptide at a cysteinyl ligand concentration of 20 mM confirmed that the stability increased with increasing length of the polypeptide. The t1/2 increased from 7 h for the tripeptide glutathione to 22 h for the hexapeptide and 35 h for the dodecapeptide (Fig. 5b and Fig. S19, ESI†). The longer peptides that consisted of repeating tripeptide units not only led to increased [2Fe–2S] stability but also to sequences with similarities to modern day ferredoxins. Many ferredoxins contain a common CxxCxxC motif that provides three of the four ligands to the iron–sulphur cluster. The final cysteine ligand is farther away in primary sequence. Repeating glutathione units give the same spacing for the first three ligands (Fig. 5c).
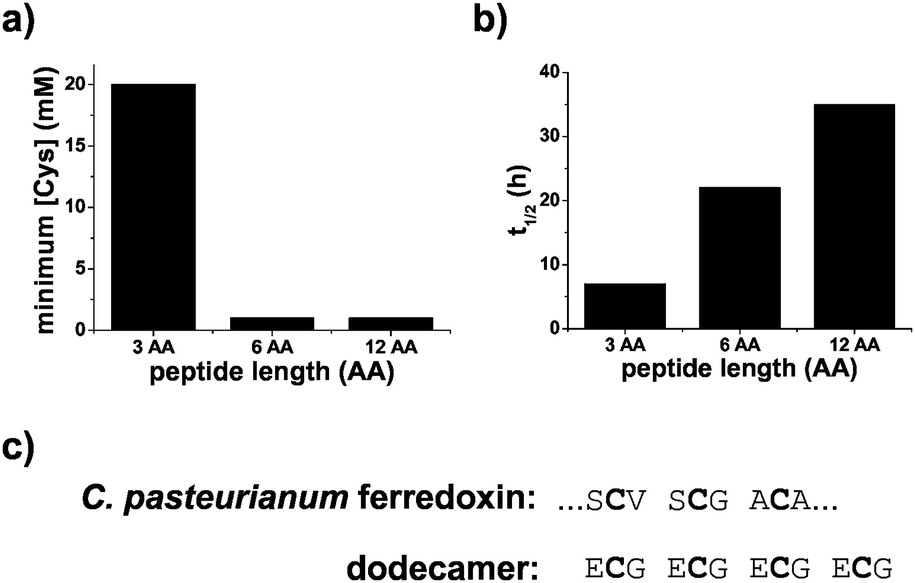 | ||
| Fig. 5 Stability of [2Fe–2S] peptides. (a) The minimum concentration of cysteinyl ligand needed to stabilize a [2Fe–2S] for glutathione and peptides containing two and four glutathione units. (b) The t1/2 of the peptide coordinated [2Fe–2S] increased as the length of the peptide increased. (c) The dodecamer contained a spacing of cysteines similar to that of the first three cysteine ligands of C. pasteurianum ferredoxin. Glutathione, hexapeptide, and dodecapeptide are abbreviated as 3 AA, 6 AA, and 12 AA, respectively. | ||
A process of duplication and accretion of abiotically synthesized peptides could have led to the emergence of contemporary protein folds.22 Putative ancient peptides identified through comparative bioinformatics analyses of modern proteins are enriched in sequences associated with the binding of nucleic acids and iron–sulphur clusters. These early peptides may have served as cofactors for catalytic RNA molecules, potentially leading to genetically encoded synthesis of proteins.22 In addition to expanding the range of chemistry available to nascent, life-like chemical systems, iron–sulphur clusters may have served as a template for the formation of longer peptide polymers. Peptide bonds between individually coordinated tripeptides, for example, could have formed through carbonyl sulphide-based chemistry23 or assisted by peptides24,25 or ribozymes with peptidyl transferase activity26 leading to hexa- and dodeca-peptides that are better able to bind [2Fe–2S] clusters. Until now, work on prebiotic peptide domains has remained theoretical. We show that what has been long hypothesized10 is supported by experimental data. A deeper investigation in the laboratory of computationally identified, putative prebiotic sequences may uncover unexplored protometabolic processes.
This work was supported by the Simons Foundation (290358), the Armenise-Harvard Foundation, and COST action CM1304. We thank Prof. J. W. Szostak for helpful comments on this manuscript and Dr. R. M. McCarrick for collecting EPR data. We thank the Center of Large Instrumentation at the University of Verona for providing access to the NMR spectrometer and the CIRCMSB and Prof. Natile for access to the mass spectrometer.
Notes and references
- P. Venkateswara Rao and R. H. Holm, Chem. Rev., 2004, 104, 527–560 CrossRef CAS PubMed.
- J. D. Kim, S. Senn, A. Harel, B. I. Jelen and P. G. Falkowski, Philos. Trans. R. Soc., B, 2013, 368, 20120257 CrossRef PubMed.
- C. L. Dupont, A. Butcher, R. E. Valas, P. E. Bourne and G. Caetano-Anollés, Proc. Nat. Acad. Sci. U. S. A., 2010, 107, 10567–10572 CrossRef CAS PubMed.
- S. E. Mulholland, B. R. Gibney, F. Rabanal and P. L. Dutton, Biochemistry, 1999, 38, 10442–10448 CrossRef CAS PubMed.
- A. Harel, Y. Bromberg, P. G. Falkowski and D. Bhattacharya, Proc. Nat. Acad. Sci. U. S. A., 2014, 111, 7042–7047 CrossRef CAS PubMed.
- F. L. Sousa, T. Thiergart, G. Landan, S. Nelson-Sathi, I. A. C. Pereira, J. F. Allen, N. Lane and W. F. Martin, Philos. Trans. R. Soc., B, 2013, 368, 20130088 CrossRef PubMed.
- G. Wachtershauser, Proc. Nat. Acad. Sci. U. S. A., 1990, 87, 200–204 CrossRef CAS.
- W. Martin, J. Baross, D. Kelley and M. J. Russel, Nat. Rev. Microbiol., 2008, 6, 805–814 CAS.
- H. Beinert, J. Biol. Inorg. Chem., 2000, 5, 2–15 CrossRef CAS PubMed.
- R. V. Eck and M. O. Dayhoff, Science, 1966, 152, 363–366 CAS.
- A. N. Lupas, C. P. Ponting and R. B. Russell, J. Struct. Biol., 2001, 134, 191–203 CrossRef CAS PubMed.
- J. D. Kim, A. Rodriguez-Granillo, D. A. Case, V. Nanda and P. G. Falkowski, PLoS Comput. Biol., 2012, 8, e1002463 CAS.
- Y. Sugiura and H. Tanaka, Biochem. Biophys. Res. Commun., 1972, 46, 335–340 CrossRef CAS PubMed.
- W. Qi, J. Li, C. Y. Chain, G. A. Pasquevich, A. F. Pasquevich and J. A. Cowan, J. Am. Chem. Soc., 2012, 134, 10745–10748 CrossRef CAS PubMed.
- W. Qi, J. Li, C. Y. Chain, G. A. Pasquevich, A. F. Pasquevich and J. A. Cowan, Chem. Commun., 2013, 49, 6313–6315 RSC.
- J. Li, S. A. Pearson, K. Fenk and J. A. Cowan, J. Biol. Inorg. Chem., 2015, 20, 1221–1227 CrossRef CAS PubMed.
- L. Skjeldal, J. L. Markley, V. M. Coghlan and L. E. Vickery, Biochemistry, 1991, 30, 9078–9083 CrossRef CAS PubMed.
- H. Beinert, R. H. Holm and E. Münck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- C. Kaszuba, P. A. Postila, O. Cramariuc, M. Sarewicz, A. Osyczka, I. Vattulainen and T. Róg, Theor. Chem. Acc., 2013, 132, 1370 CrossRef.
- B. M. Bode and M. S. Gordon, J. Mol. Graphics Modell., 1998, 16, 133–138 CrossRef CAS PubMed.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry: The First Forty Years, ed. C. E. Dykstra, G. Frenking and G. E. Scuseria, Elsevier B.V., Amsterdam, 2005, pp. 1167–1189 Search PubMed.
- V. Alva, J. Soding and A. N. Lupas, eLife, 2015, 4, e09410 Search PubMed.
- L. Leman, L. Orgel and M. R. Ghadiri, Science, 2004, 306, 283–286 CrossRef CAS PubMed.
- M. Gorlero, R. Wieczorek, K. Adamala, A. Giorgi, M. E. Schininà, P. Stano and P. L. Luisi, FEBS Lett., 2009, 583, 153–156 CrossRef CAS PubMed.
- K. Adamala and J. W. Szostak, Nat. Chem., 2013, 5, 495–501 CrossRef CAS PubMed.
- B. Zhang and T. R. Cech, Nature, 1997, 390, 96–100 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Tables S1–S3 and Fig. S1–S19. See DOI: 10.1039/c6cc07912a |
| This journal is © The Royal Society of Chemistry 2016 |