
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II)-catalyzed C–H functionalizations on benzoic acids with aryl, alkenyl and alkynyl halides by weak-O-coordination†
Ruhuai
Mei‡
,
Cuiju
Zhu‡
and
Lutz
Ackermann
 *
*
Institut für Organische und Biomolekulare Chemie, Georg-August-Universität, Tammannstraße 2, 37077 Goettingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 18th October 2016
Abstract
C–H arylations of weakly coordinating benzoic acids were achieved by versatile ruthenium(II) catalysis with ample substrate scope. Thus, user-friendly ruthenium(II) biscarboxylate complexes modified with tricyclohexylphosphine enabled C–H functionalizations with aryl electrophiles. The unique versatility of the ruthenium(II) catalysis manifold was reflected by facilitating effective C–H activations with aryl, alkenyl and alkynyl halides.
Transformations of unactivated C–H bonds have emerged as an attractive alternative to conventional cross-coupling approaches, enabling step-economical biaryl syntheses with reduced by-product formation.1 Major progress has been accomplished by means of ruthenium(II)-catalyzed reactions with easily accessible electrophilic2–6 aryl halides,7–10 with transformative applications in material sciences,11 as well as agrochemical12 and pharmaceutical industries,13,14 among others.7,8 Despite these undisputable advances, ruthenium(II)-catalyzed C–H arylations with organic electrophiles continue to be limited to strongly coordinating nitrogen-containing directing groups,7,8 which are difficult to remove15 or modify (Fig. 1a).16 Within our ongoing program on ruthenium-catalyzed C–H functionalizations,17,18 we have now developed the unprecedented ruthenium(II)-catalyzed C–H arylations of benzoic acids,19 on which we report herein (Fig. 1b). The key to success was represented by using a tricyclophosphine-derived ruthenium(II) complex, which we have previously developed for C–H functionalizations guided by strong N-coordination.20 Notable features of our approach include (i) first ruthenium-catalyzed C–H arylations of weakly O-coordinating21,22 benzoic acids, (ii) mechanistic insights on facile carboxylate-assisted C–H activation, and (iii) a versatile ruthenium(II) catalysis regime that set the stage for expedient C–H transformations with challenging aryl, alkenyl and alkynyl halides.
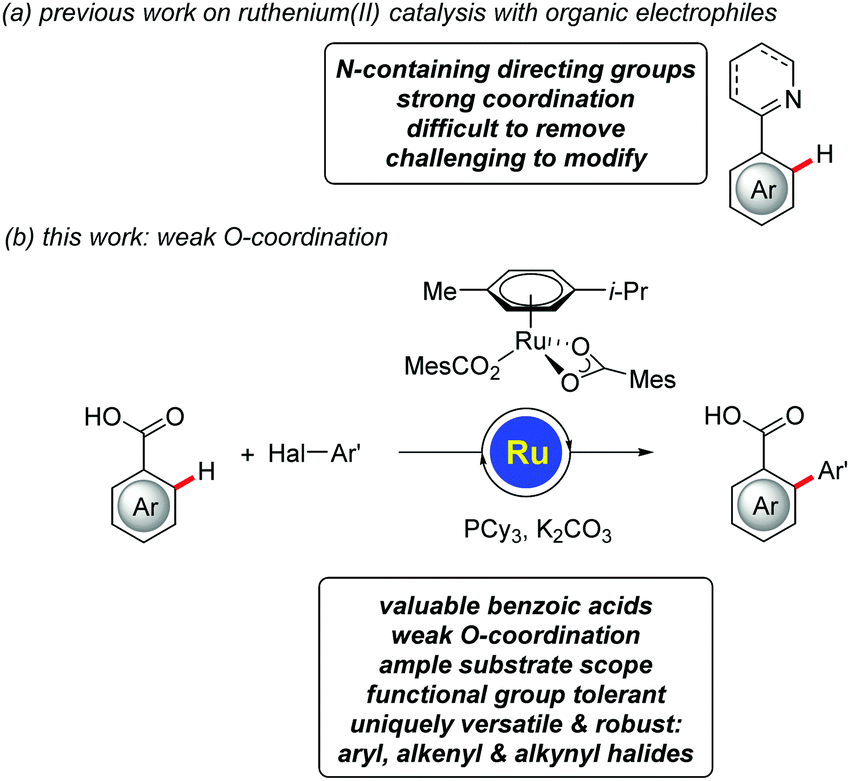 | ||
| Fig. 1 Ruthenium(II)-catalyzed C–H arylation by weak coordination. | ||
At the outset of our studies, we explored reaction conditions for the envisioned ruthenium(II)-catalyzed C–H arylation of weakly O-coordinating benzoic acids 1a (Table 1 and Table S1 in the ESI†).23 While typical phosphine or N-heterocyclic carbene ligands fell short in providing access to any arylated benzoic acid products (entries 1–8), a PCy3-derived catalyst – previously exploited for strongly N-coordinating 1,2,3-triazoles20 – enabled the challenging C–H arylation process (entries 9 and 10). It is noteworthy that the well-defined [RuCl2(PCy3)(p-cymene)] was also identified as a user-friendly single component catalyst, allowing for the preparation of the ortho-arylated benzoic acid 3aa with comparable levels of efficacy (entry 11). The catalytic performance was further significantly improved by exploiting carboxylate24 assistance with the aid of the well-defined ruthenium(II)biscarboxylate complex 425 (entries 12–14).
|
|
|||
|---|---|---|---|
| Entry | [Ru] | Ligand | 3aa (%) |
| a Reaction conditions: 1a (0.50 mmol), 2a (0.75 mmol), [Ru] (10 mol%), additive (10 mol%), K2CO3 (2.0 equiv.), NMP (2.0 mL), 120 °C, 16 h; then K2CO3 (3.0 equiv.), MeI (5.0 equiv.), MeCN (3.0 mL), 50 °C, 2 h. b Yields of isolated product; in parentheses: GC conversion after esterification with 1,3,5-trimethoxybenzene as the internal standard. c Without K2CO3. d DMPU (2.0 mL) as the solvent. e DMA (2.0 mL) as the solvent. | |||
| 1 | [RuCl2(p-cymene)]2 | — | (11) |
| 2 | [RuCl2(p-cymene)]2 | IPrHCl | (<5) |
| 3 | [RuCl2(p-cymene)]2 | IMesHCl | (<5) |
| 4 | [Ru(MesCO2)2(p-cymene)] | X-Phos | (7) |
| 5 | [RuCl2(p-cymene)]2 | DavePhos | (<5) |
| 6 | [RuCl2(p-cymene)]2 | JohnPhos | (8) |
| 7 | [RuCl2(p-cymene)]2 | (tBu)2POH | (20) |
| 8 | [RuCl2(p-cymene)]2 | P(tBu)3 | (22) |
| 9 | [RuCl2(p-cymene)]2 | PCy3 | 81 |
| 10 | [RuCl2(p-cymene)]2 | PCy3 | —c |
| 11 | [RuCl2(PCy3)(p-cymene)] | — | 75 |
| 12 | [Ru(MesCO2)2(p-cymene)] (4) | PCy3 | (32)d |
| 13 | [Ru(MesCO2)2(p-cymene)] (4) | PCy3 | 54e |
| 14 | [Ru(MesCO2)2(p-cymene)] (4) | PCy3 | 87 |

With the optimized catalyst in hand, we probed its versatility in the C–H arylation of differently substituted aryl halides 2 (Scheme 1). Here, a representative set of synthetically meaningful functional groups, such as halides, activated alkenes, esters or enolizable ketones, was well tolerated by the optimized catalyst at different positions of the organic electrophile 2. Moreover, the robustness of the ruthenium(II) catalyst was reflected by efficiently converting both electron-deficient as well as more demanding electron-rich aryl halides 2.
 | ||
| Scheme 1 C–H activation of weakly O-coordinating benzoic acid 1a with aryl bromides 2. | ||
Subsequently, we probed the scope of viable benzoic acids in the ruthenium(II)-catalyzed C–H arylation manifold (Scheme 2). Thus, we were delighted to observe that various weakly-coordinating acids 1 could be converted with high catalytic efficacy and excellent positional selectivity by the phosphine-modified biscarboxylate complex 4. Importantly, the versatile ruthenium(II) catalyst was not restricted to arenes. Indeed, the biscarboxylate complex 4 also allowed for the site-selective C–H arylation of synthetically useful indole 1n. Interestingly, the ligand JohnPhos outcompeted PCy3 in the heteroarene diversification.
 | ||
| Scheme 2 C–H arylation of benzoic acids 1 by ruthenium(II) catalysis. a Isolated yield of the mono-arylated product. b With ArI instead of 2a. c 9% of the di-arylated product isolated. d JohnPhos (10 mol%) instead of PCy3. | ||
In consideration of the unique efficiency of the ruthenium(II) catalysis regime, we became intrigued by rationalizing its mode of action. To this end, intermolecular competition experiments revealed electron-deficient aryl bromides to react preferentially (Scheme 3).
 | ||
| Scheme 3 Intermolecular competition experiment. | ||
The challenging nature of the C–H arylation with weakly coordinating benzoic acids became apparent by an intermolecular competition experiment between benzoic acid 1h and arene 5d displaying the strongly N-coordinating 1,2,3-triazole (Scheme 4).
 | ||
| Scheme 4 Intermolecular competition experiment. | ||
Moreover, we observed a significant H/D scrambling upon the addition of an isotopically labeled cosolvent under otherwise identical reaction conditions. The deuterium incorporation in the reisolated substrate [D]n–1o and product [D]n–3oa is supportive of a reversible C–H metalation event (Scheme 5).
 | ||
| Scheme 5 Facile C–H arylation in the presence of isotopically labeled cosolvent. | ||
The well-defined ruthenacycle 7, that we had previously employed for oxidative alkyne annulations,26 was shown to be catalytically competent (Scheme 6), being indicative of an organometallic mode of C–H activation.
 | ||
| Scheme 6 Ruthenacycle 7 for C–H arylation. | ||
Finally, the unique versatility of the ruthenium(II) catalysis was illustrated by the phosphine-modified catalyst 4 enabling the unprecedented olefination and alkynylation of benzoic acids 1 by alkenyl and alkynyl halides 8 and 10, respectively (Scheme 7). Both types of C–H functionalization occurred by weak O-coordination with excellent levels of positional selectivities, thereby providing access to ortho-alkenylated benzoic acids 9 and phthalide27,28 derivatives 11 – key structural motifs of naturally occurring compounds.29
 | ||
| Scheme 7 Weak O-coordination for (a) C–H alkenylation and (b) C–H alkynylation. | ||
In summary, we have developed the first ruthenium(II)-catalyzed C–H functionalization of weakly O-coordinating arenes with organic halides. Thus, a versatile phosphine-modified30 ruthenium(II) biscarboxylate catalyst enabled C–H arylations of benzoic acids with excellent positional selectivity and ample scope. The facile C–H ruthenation manifold enabled the direct arylation of aromatic and heteroaromatic carboxylic acids. Furthermore, the unique synthetic utility of the ruthenium(II) catalysis regime also set the stage for site-selective C–H olefinations and C–H alkynylations of benzoic acids under otherwise identical reaction conditions. Further studies on ruthenium(II)-catalyzed C–H functionalization by weak coordination are ongoing in our laboratories and will be reported in due course.
Generous support by the European Research Council under the European Community's Seventh Framework Program (FP7 2007–2013)/ERC Grant agreement no. 307535, and the CSC (fellowships to M. R. and C. Z.) is gratefully acknowledged.
References
- Representative reviews on C–H activation: (a) J. G. Kim, K. Shin and S. Chang, Top. Organomet. Chem., 2016, 55, 29–51 CrossRef; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (c) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (d) N. Kuhl, N. Schroeder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (e) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (f) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Organic Chemistry Frontiers, 2014, 1, 843–895 RSC; (g) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (h) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (i) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (j) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (k) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed , and references cited therein.
- C. Sollert, K. Devaraj, A. Orthaber, P. J. Gates and L. T. Pilarski, Chem. – Eur. J., 2015, 21, 5380–5386 CrossRef CAS PubMed.
- J. Hubrich, T. Himmler, L. Rodefeld and L. Ackermann, Adv. Synth. Catal., 2015, 357, 474–480 CrossRef CAS.
- (a) R. K. Chinnagolla, A. Vijeta and M. Jeganmohan, Chem. Commun., 2015, 51, 12992–12995 RSC; (b) R. K. Chinnagolla and M. Jeganmohan, Org. Lett., 2012, 14, 5246–5249 CrossRef CAS PubMed.
- F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698–1699 CrossRef CAS PubMed.
- K. Kitazawa, T. Kochi, M. Sato and F. Kakiuchi, Org. Lett., 2009, 11, 1951–1954 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- L. Ackermann and R. Vicente, Top. Curr. Chem., 2010, 292, 211–229 CrossRef CAS PubMed.
- S. Oi, E. Aizawa, Y. Ogino and Y. Inoue, J. Org. Chem., 2005, 70, 3113–3119 CrossRef CAS PubMed.
- L. Ackermann, Org. Lett., 2005, 7, 3123–3125 CrossRef CAS PubMed.
- W. Lu, J. Kuwabara and T. Kanbara, Macromol. Rapid Commun., 2013, 34, 1151–1156 CrossRef CAS PubMed.
- J. Hubrich, T. Himmler, L. Rodefeld and L. Ackermann, ACS Catal., 2015, 5, 4089–4093 CrossRef CAS.
- M. Seki, Org. Process Res. Dev., 2016, 20, 867–877 CrossRef CAS.
- L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS.
- L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154–1157 CrossRef CAS PubMed.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed.
- L. Ackermann, Synlett, 2007, 507–526 CrossRef CAS.
- For arylations of benzoic acids catalyzed by metals other than ruthenium, see: (a) L. Huang, D. Hackenberger and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 12607–12611 CrossRef CAS PubMed; (b) C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418–3421 CrossRef CAS PubMed; (c) Z. Wu, S. Chen, C. Hu, Z. Li, H. Xiang and X. Zhou, ChemCatChem, 2013, 5, 2839–2842 CrossRef CAS; (d) C. Arroniz, J. G. Denis, A. Ironmonger, G. Rassias and I. Larrosa, Chem. Sci., 2014, 5, 3509–3514 RSC; (e) C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910–913 CrossRef CAS PubMed; (f) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S. P. Breazzano, L. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510–3511 CrossRef CAS PubMed; (g) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed , and references cited therein.
- L. Ackermann, R. Born and R. Vicente, ChemSusChem, 2009, 546–549 CrossRef CAS PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- For detailed information, see the ESI†.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed.
- S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS PubMed.
- H. Zhao, T. Zhang, T. Yan and M. Cai, J. Org. Chem., 2015, 80, 8849–8855 CrossRef CAS PubMed.
- (a) A. Bechtoldt, C. Tirler, K. Raghuvanshi, S. Warratz, C. Kornhaaß and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 264–267 CrossRef CAS PubMed; (b) L. Ackermann and J. Pospech, Org. Lett., 2011, 13, 4153–4155 CrossRef CAS PubMed.
- J. J. Beck and S.-C. Chou, J. Nat. Prod., 2007, 70, 891–900 CrossRef CAS PubMed.
- The electron-rich phosphine ligand PCy3 is proposed to facilitate the C–H functionalization on the weakly coordinating benzoic acid.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR spectra for products. See DOI: 10.1039/c6cc07773k |
| ‡ RM and CZ contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |