
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis of nitroaryl acetylenes from acetylenes and nitroarenes via oxidative nucleophilic substitution of hydrogen†
Robert
Bujok
and
Mieczysław
Mąkosza
*
Institute of Organic Chemistry Polish Academy of Sciences, ul. Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: icho-s@icho.edy.pl
First published on 28th September 2016
Abstract
Acetylenic carbanions add to nitroarenes (dinitrobenzenes, nitropyridines, etc.) to form σH-adducts that are subsequently oxidized by DDQ according to the oxidative nucleophilic substitution of hydrogen (ONSH) pathway to give nitroaryl acetylenes.
Introduction of acetylenic substituents into aromatic rings is of great interest in modern organic synthesis, because the presence of such fragments adds interesting physicochemical properties1 and particularly because they can be converted into a variety of functionalities. Of particular value for organic synthesis are interactions of the acetylenic moieties in aromatic rings with vicinal substituents to form heterocyclic or carbocyclic rings.2 For instance, transition metal catalyzed cyclization reactions of ortho-amino-arylacetylenes are widely used for synthesis of indoles.3 The most general method of introduction of acetylenic substituents into aromatic rings is the Sonogashira reaction – transition metal catalyzed substitution of halogens in aromatic rings by terminal alkynes and arylacetylenes. The reaction is of general character and, as a rule, proceeds with high yield; however, the products are contaminated with traces of transition metals that are often undesirable.4
There are several examples published on replacement of halogens in aromatic rings by acetylenic carbanions without using transition metal catalysts; however, the starting arenes should still contain halogens.5 Recently, a very interesting way of introduction of acetylenic substituents into nitroarenes was reported via SNAr of fluorine in ortho- and para-fluoronitroarenes by enolates α-(2-benzothiazyl)ketones followed by Julia type olefination that proceeds via Smiles rearrangement.6
It should be mentioned that lithium and potassium acetylenides add to electron deficient azines such as 1,2,4-triazines and triazine oxides, etc. Further conversion of the σH-adducts proceeds via oxidation or rearrangement to form acetylenyl or styrylazines.7
All reported methods of introduction of acetylenic substituents into aromatic rings consist of replacement of halogens: transition metal catalyzed replacement of I, Br, and Cl by terminal acetylenes – Sonogashira reaction and SNAr of fluorine in fluoronitrobenzenes by acetylenic carbanions. These are not atom economic methods, because the halogen atom introduced into the aromatic ring in the first step of the synthesis of nitroaryl acetylenes is substituted by the acetylenic moiety subsequently.
Herein, we report an atom economical method for synthesis of nitroaryl acetylenes from acetylenes and nitroarenes via oxidative nucleophilic substitution of hydrogen, ONSH, with acetylenic carbanions.
ONSH consists of addition of nucleophiles such as OH−, NH3, ArNH2 and particularly carbanions to nitroarenes at positions occupied by hydrogen to form σH adducts that are subsequently oxidized by external oxidants to form products of oxidative nucleophilic substitution of hydrogen. It should be stressed that addition of nucleophiles to halonitroarenes proceeds faster at positions occupied by hydrogen than at those occupied by halogens; hence ONSH proceeds also in halonitrobenzenes.8 Carbanions are sensitive to oxidation; thus ONSH proceeds efficiently, provided the addition equilibrium is shifted towards the adducts. The equilibrium depends on the electrophilicity of nitroarenes, nucleophilicity of carbanions and the conditions. Carbanions should be in the form of loose ion-pairs and the reaction should be carried out at low temperature. Taking into account the versatility of ONSH in nitroarenes with a variety of nucleophiles, we expect that such reaction should be applicable for acetylenide anions. For generation of acetylenide carbanions the most convenient base–solvent system is n-BuLi in THF. Since for the addition to nitroarenes carbanions should be in the form of loose ion-pairs, HMPA was added to the reaction mixture.
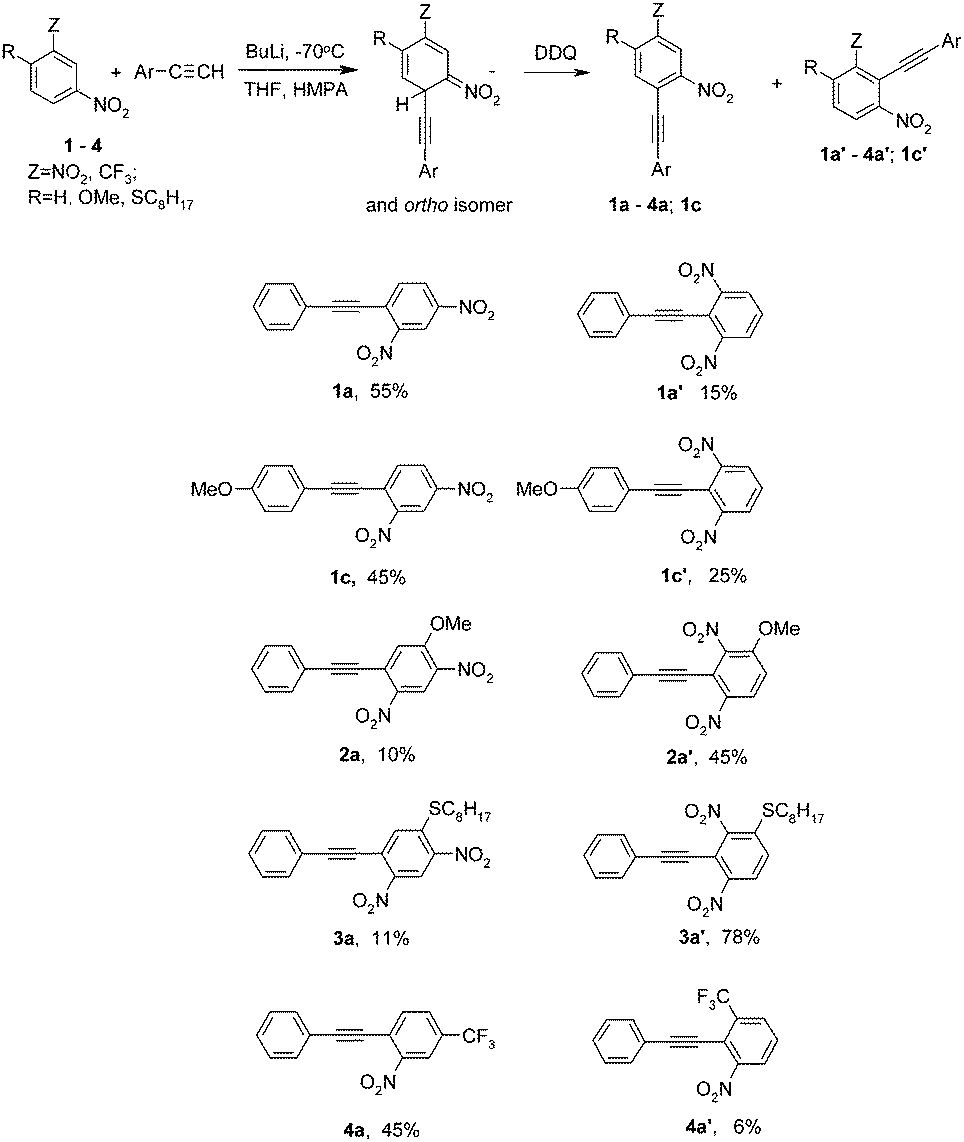
The first attempts at reacting nitrobenzene and p-chloronitrobenzene with lithium phenylacetylenide in THF in the presence of 1 equivalent of HMPA (calculated on used BuLi) at −70 °C with DDQ as an oxidant gave negative results. No expected products were produced. This was, apparently, because acetylenide carbanions, although the acetylenes are weak CH acids (pKa of phenylacetylene 28.8),9 are surprisingly weak nucleophiles and with relatively weak electrophiles such as these nitroarenes do not form σH adducts in a degree sufficient to afford ONSH. On the other hand, with a much stronger electrophile, m-dinitrobenzene, the reaction proceeded satisfactorily, giving a mixture of two isomeric 2,4- and 2,6-dinitrotolanes 1a and 1a′ (isolated yields 55% and 15% respectively). A linear acetylenic carbanion can also add at highly sterically hindered position 2 of m-dinitrobenzene. It should be noted that the methylenic carbanion of chloromethyl phenyl sulfone adds to 1 exclusively at position 4.10
The preliminary results indicated that for ONSH with carbanions of phenylacetylene and other acetylenes, nitroarenes of high electrophilic activities should be used. For selection of the appropriate, sufficiently active nitroarenes, we have used reported values of the electrophilic activities of nitroarenes and nitroheteroarenes determined by measurements of relative rates of the addition of carbanions of chloromethyl phenyl sulfone to a variety of nitroarenes and nitroheteroarenes as a rate limiting step of the vicarious nucleophilic substitution of hydrogen, VNS.11 The electrophilic activities are quantitatively expressed as the relation of rates of the addition of this carbanion to various nitroarenes to the rate of the addition at the ortho positions of nitrobenzene.11
On the basis of this criterion we have selected a series of highly active nitroarenes. However, under the conditions used for m-dinitrobenzene 1, 1-nitronaphtalene 5 (e = 4600) and m-trifluoromethylnitrobenzene 4 (e = 5000) did not react with lithium phenylacetylenide. Probably an acetylenide carbanion was too much associated with a lithium cation to react with less active nitroarenes than m-dinitrobenzene. To increase the activity of the carbanion, HMPA was used not as a reagent, but as a co-solvent. Indeed, under these conditions,12 the desired reactions proceed satisfactorily. The expected products were obtained in moderate yields (42% in the case of 1-nitronaphtalene 5 and 51% for m-trifluoromethyl-nitrobenzene 4; a mixture of isomers 4a and 4a′ was formed in the last case). These are the first examples of ONSH reactions in nitrobenzenes with carbanions formed from acetylenes. Unfortunately, even when HMPA was used as a co-solvent, p-chloronitrobenzene (e = 130) was still inactive under the reaction conditions and the corresponding product was not formed. The results of the ONSH reaction of acetylenes with nitroarenes are shown in Tables 1 and 2.
|

|
The reaction of lithium phenylacetylenide with 2,4-dinitroanisole 2 (e = 9000) gave three products of substitution of hydrogen at position 5-, 2a (10%); 3-, 2a′ (45%) and the methoxy group 1a (17%). Nucleophilic substitution of the methoxy group in 2 is a common process that often proceeds faster than SNAr of chlorine.13 Preferred addition at position 3- is due to the conjugation of the electron-donating methoxy group with the nitro groups. A similar effect of the conjugation on the orientation of the nucleophile addition to 2,4-dinitroanisole 2, 2,4-dinitrophenolate and 2,4-dinitro-N-methylaniline was observed in the vicarious nucleophilic substitution (VNS) reaction with the methylenic carbanion of chloromethyl phenyl sulfone.
Due to strong conjugation in the phenolate and aniline, the VNS reaction in these nitroarenes proceeds exclusively at position 3-, whereas conjugation in the anisole is much weaker, so VNS proceeds at position 5-.14
A very good yield was achieved in the reaction of phenylacetylene with sulfide 3 (78% of isomer 3a′ and 11% of isomer 3a). It is an important result, because it is known that sulfides inhibit transition metal catalyzed reactions.
The ONSH with acetylenide carbanions proceeds particularly efficiently in 3-nitropyridine 6 (e = 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and 2-nitro-5-methoxy-pyridine 7 (e = 17000), which are much more active than nitroarenes 4 and 5. In both the nitropyridines the substitution proceeds selectively in one position. In the case of 3-nitropyridine 6 the reaction proceeds at position 4-, whereas substitution in 7 occurs selectively at position 2-. As in the case of 2, the orientation in 7 is governed by the conjugation of the methoxy and the nitro group (Scheme 1). Such an effect on orientation was observed also in the VNS in methoxynitropyridines with methylenic α-chloro carbanions.15
000) and 2-nitro-5-methoxy-pyridine 7 (e = 17000), which are much more active than nitroarenes 4 and 5. In both the nitropyridines the substitution proceeds selectively in one position. In the case of 3-nitropyridine 6 the reaction proceeds at position 4-, whereas substitution in 7 occurs selectively at position 2-. As in the case of 2, the orientation in 7 is governed by the conjugation of the methoxy and the nitro group (Scheme 1). Such an effect on orientation was observed also in the VNS in methoxynitropyridines with methylenic α-chloro carbanions.15
 | ||
| Scheme 1 | ||
A series of arylacetylenes and TMS-acetylene in the reaction with 7 gave expected products 7a–7e in good yields (72–83%). Regarding the acetylene moiety, this method is of general character. Good results were obtained for phenylacetylenes with a strong electron donor (–OMe), a weak electron donor (–Me) and a weak electron acceptor group (–F). The TMS group in trimethylsilylacetylene is resistant under conditions of the reaction and the expected product 7e was obtained in good yield (68%). On the other hand, the yield of the reaction with n-butyl acetylene under the standard conditions was only moderate (40%). It appears that it was because alkyl acetylenes under basic conditions can rearrange into allenes. Indeed, when the generated lithium acetylenide was immediately mixed with 7, the yield of 7e was somewhat higher (54%; GC yield).
In conclusion, we have developed a transition metal free, atom economical method for synthesis of nitroaryl acetylenes from acetylenes and nitroarenes, via direct replacement of hydrogen. The method consists of addition of lithium acetylenides to electron-deficient nitroarenes carbo- and heterocyclic to form σH-adducts, which are subsequently oxidized by DDQ, giving nitroaryl-acetylenes. Regarding the acetylene moiety, this method is of general character. On the other hand, the reaction is limited to highly electrophilic nitroarenes; nevertheless, it is the simplest way of introduction of an acetylenic moiety into nitroaromatic rings.
This work was supported by the National Science Centre (grant UNO 2014/15/B/ST5/0218).
Notes and references
- (a) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef CAS PubMed; (b) M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- (a) F. Denes, A. Perez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366 CrossRef CAS PubMed; (b) A. Kondoh, H. T. Q. Tran, K. Kimura and M. Terada, Chem. Commun., 2016, 52, 5726 RSC; (c) T. Aggarwal, S. Kumar and A. K. Verma, Org. Biomol. Chem., 2016, 14, 7639 RSC.
- (a) R. Vincente, Org. Biomol. Chem., 2011, 9, 6469 RSC; (b) M. Michalska and K. Grela, Synlett, 2016, 599 CAS; (c) S. Song, M. Huang, W. Li, X. Zhu and Y. Wan, Tetrahedron, 2015, 71, 451 CrossRef CAS; (d) C. B. Lavery, R. McDonald and M. Stradiotto, Chem. Commun., 2012, 48, 7277 RSC; (e) G. Zeni and R. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS PubMed; (f) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (g) R. Karmahar, A. Suneja and V. D. Singh, Org. Lett., 2016, 18, 2636 CrossRef PubMed; (h) P. Li, Y. Weng, X. Xu and X. Cui, J. Org. Chem., 2016, 81, 3994 CrossRef CAS PubMed; (i) X. Yuan, X. Wu, S. Dong and J. Ye, Org. Biomol. Chem., 2016, 14, 7447 RSC.
- (a) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 CrossRef CAS PubMed; (b) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (c) A. C. Shaikh, D. W. Shinde and N. T. Patil, Org. Lett., 2016, 18, 1056 CrossRef CAS PubMed; (d) B. Miao, S. Li, G. Li and S. Ma, Org. Lett., 2016, 18, 2556 CrossRef CAS PubMed.
- (a) P. L. DeRoy, S. Surprenant, M. Bertrand-Laperle and C. Yoakim, Org. Lett., 2007, 5, 2741 CrossRef PubMed; (b) R. Luque and D. J. Macquarrie, Org. Biomol. Chem., 2009, 7, 1627 RSC.
- B. Prüger, G. E. Hofmeister, C. B. Jacobsen, D. G. Alberg, M. Nielsen and K. A. Jørgensen, Chem. – Eur. J., 2010, 16, 3783 CrossRef PubMed.
- (a) A. M. Prokhorov, M. Mąkosza and O. N. Chupakhin, Tetrahedron Lett., 2009, 50, 1444 CrossRef CAS; (b) F. I. Carroll, S. V. Kotturi, H. A. Navarro, A. W. Mascarella, B. P. Glimour, F. L. Smith, B. H. Gabra and W. L. Dewey, J. Med. Chem., 2007, 50, 3388 CrossRef CAS PubMed; (c) A. F. Khasanow, D. S. Kopchuk, I. S. Kovalov, O. S. Taniya, G. V. Zyryanov, V. L. Rusinov and O. N. Chupakhin, Mendeleev Commun., 2015, 25, 332 CrossRef.
- (a) M. Mąkosza, in Arene Chemistry: Reaction Mechanisms and Methods for Aromatic Compounds, ed. J. Mortier, Wiley, ch. 11, 2016, p. 269 Search PubMed; (b) M. Mąkosza, Chem. – Eur. J., 2014, 20, 5536 CrossRef PubMed; (c) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855 RSC; (d) E. V. Malykhin, G. A. Kolesnichenko and V. D. Shteingarts, Zh. Org. Khim., 1985, 21, 1150 CAS; (e) H. C. Van der Plas and M. Woźniak, Croat. Chem. Acta, 1986, 59, 33 CAS; (f) V. V. Khutorianski, M. Sonawane, M. Posta, B. Klepetarova and P. Beier, Chem. Commun., 2016, 52, 7237 RSC; (g) M. Mąkosza and K. Staliński, Chem. – Eur. J., 1997, 3, 2025 CrossRef; (h) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665 CrossRef CAS.
- F. G. Bordwell, G. E. Drucker, N. H. Andersen and A. D. Denniston, J. Am. Chem. Soc., 1986, 108, 7310 CrossRef CAS.
- M. Mąkosza, J. Goliński and J. Baran, J. Org. Chem., 1984, 49, 1488 CrossRef.
- (a) S. Błażej and M. Mąkosza, Chem. – Eur. J., 2008, 14, 11113 CrossRef PubMed; (b) F. Seelinger, S. Błażej, S. Bernhardt, M. Mąkosza and H. Mayr, Chem. – Eur. J., 2008, 14, 6108 CrossRef PubMed.
- To a solution of acetylene (3.0 mmol) in dry THF (7 mL) and HMPA (5 mL) at −70 °C, 2.5 M BuLi in hexane (1.2 mL, 3.0 mmol) was added in 4 min and the mixture was stirred at −70 °C for 30 min. A solution of the nitroarene 2–7 (1.5 mmol) in THF (2.0 mL) was added and the resultant mixture was stirred for 1 h at −70 °C. A solution of 0.68 g (3.0 mmol) of DDQ in THF (2.0 mL) was added and the mixture was stirred at −70 °C for 20 min and then 1 h at RT. After evaporation of THF, the residue was subjected to column chromatography (SiO2, hexane/AcOEt).
- M. J. Miller, Aromatic Nucleophilic Substitution, Elsevier, 1968, p. 166 Search PubMed.
- (a) M. Mąkosza and S. Ludwiczak, J. Org. Chem., 1984, 49, 4562 CrossRef; (b) M. Mąkosza, S. Voskresensky, M. Białecki and A. Kwast, Pol. J. Chem., 1999, 73, 1969 Search PubMed.
- (a) J. M. Bakke, E. J. Andreassen, I. Stetvold and H. Svensen, Org. Biomol. Chem., 2004, 4, 2671 Search PubMed; (b) M. Mąkosza, B. Chylińska and B. Mudryk, Liebigs Ann. Chem., 1984, 8 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07475h |
| This journal is © The Royal Society of Chemistry 2016 |