
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution structures of alkali metal cyclopentadienides in THF estimated by ECC-DOSY NMR-spectroscopy (incl. software)†
Sebastian
Bachmann
a,
Björn
Gernert
b and
Dietmar
Stalke
*a
aInstitut für Anorganische Chemie, Georg-August-Universität, Tammannstraße 4, D-37077, Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
bInstitut für Betriebssysteme und Rechnerverbund, TU Braunschweig, Mühlenpfordstraße 23, Braunschweig, D-38106, Germany
First published on 7th October 2016
Abstract
In this paper we present the aggregational motifs of the widely used alkali-metal cyclopentadienides (CpLi, CpNa, CpK, CpRb, CpCs) in THF-d8 solution estimated by ECC-DOSY NMR spectroscopy. They form monomeric contact ion pairs (CIPs) in THF-d8 solution, whereas in NH3 solvent-separated ion pairs (SSIPs) are observed. The applicability of ECC-DOSY is further advanced by introducing ECC-MW estimation software.
Alkali metal cyclopentadienide (CpM) derivatives are among the most widely used starting materials in organometallic synthesis.1 They can be used to synthesise a variety of different sandwich or half-sandwich d-block organometallics through transmetallation or salt elimination, which in turn can be used e.g. as polymerisation catalysts.2–4 Ferrocene, which was synthesised by Kealy and Pauson in 1951 is often described as the first metallocene, but alkali metal cyclopentadienides CpMs were indeed synthesised more than 50 years earlier (Thiele, 1900).5,6 The solubility of CpM compounds has to be described as poor in hydrocarbons as well as in ethers. THF is the only solvent that provides reasonable concentrations in solution. High melting points and low volatilities also underline their salt-like character.7,8 Crystal structure analysis shows that the donor free compounds form polymeric chains that are linear for [CpLi]∞ and [CpNa]∞ but bent for all higher homologues (for CpRb there are ambivalent structures, Scheme 1).9–11 Various solvation states account for various excerpts from the solid state structures12 like for example [Li(NH3)4]+[Cp]−,13 [(TMEDA)LiCp],14 [(H3N)3NaCp],15 [(H3N)3LiCpLiCp],13 [Ph4P]+[Cp2Li]−,16 or [Ph4P]+[Cp3Cs2]−.17
 | ||
| Scheme 1 Solid state structures of parent [CpM]∞. | ||
Most of the crystal structures have to be classified as contact ion pairs (CIP), but there are a few examples of solvent-separated ion pairs (SSIPs) preferentially generated by the use of ammonia or crown ethers.13,18 Aggregation of organometallic molecules in solution determines not only their reactivity but can also shed light on reaction mechanisms, and hence provide a handle to improve e.g. yields and/or selectivities. Though crystal structure analysis can elucidate the molecular bonding situation,15 in many cases it differs quite substantially from solution structures, which in turn can vary in different solvents considerably. Therefore, the absolute size of molecules in solution has been of interest for a long time. In 1992 Johnson and Morris introduced an NMR spectroscopic method to tackle this problem: DOSY (Diffusion Ordered SpectroscopY).19 This technique correlates chemical shift information with the self-diffusion coefficient D of the compound. Li and Williard et al. pioneered DOSY NMR spectroscopy for small reactive organometallic molecules by addition of at least three internal references to one NMR sample to get an internal calibration curve (ICC).20 In the last decades many methods have been proposed to link diffusion coefficients either to molecule sizes or molecular weights (MWs).21–25 Recently, our group developed and published an advanced method.26 We established power law based external calibration curves (ECCs) together with normalised diffusion coefficients to estimate the MWs of organometallic compounds via DOSY in solution with much improved accuracy compared to previous approaches.27–30 We were also able to extend the scope of this method towards other commonly used solvents e.g. DMSO-d6.31 In the current study samples were prepared by addition of internal references to CpMs in order to normalise the diffusion coefficients of these analytes (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Dx,norm).32 This way MWs can be estimated independently of NMR spectrometer properties and differences in temperature or viscosity en route to estimate MWs for the solution structures of CpLi, CpNa, CpK, CpRb and CpCs in THF-d8. CpLi was also analysed in ammonia solution.
Dx,norm).32 This way MWs can be estimated independently of NMR spectrometer properties and differences in temperature or viscosity en route to estimate MWs for the solution structures of CpLi, CpNa, CpK, CpRb and CpCs in THF-d8. CpLi was also analysed in ammonia solution.
In THF the 1H- and 13C-NMR spectra of all alkali metal cyclopentadienides show only a single resonance (see ESI†). The signal tends to be shifted towards higher field when descending Group 1. 7Li and 133Cs NMR resonances for CpLi and CpCs, respectively, were found to be in accordance with literature.34,35 The 23Na NMR resonance (−28 to −31 ppm) is shifted towards lower field compared to solid state MAS NMR findings for CpNa and [CpNa·THF] and can be shifted even lower upon cooling.12 The same was observed for the 7Li NMR signal, which shifts and also broadens upon cooling. In 1990 it has been reported by Paquette et al. that CpLi undergoes a fast exchange process between monomeric and a “sandwich” dimeric species, which results in the splitting of the 6Li signal at lower temperatures, but they could not “tell whether the monomer–dimer equilibrium of “CpLi” is shifted to either side at room temperature”.36 The neat lithocene anion [Cp2Li]− is characterised triply in the solid state.13,16,37 We recorded spectra in THF solution down to −100 °C and could not observe any splitting of the 7Li NMR signal and at −105 °C CpLi precipitates. In addition, DOSY-NMR-spectroscopy was used to estimate MWs with ECCs. As shown before,26 molecular shapes are quite important to accurately interpret diffusion data. We found that for most organometallic compounds the dissipated spheres and ellipsoids (DSE) calibration curve is the most appropriate, especially for lithiated compounds.27 Therefore, all MWs (MWdet) of CpMs in THF were estimated via the DSE-ECC, if not stated otherwise. It seems also mandatory to calculate molecular densities (MDw) to foresee whether or not proposed structures might evoke deviations in the ECC-MW estimation.38 For all herein proposed aggregates, however, such errors due to higher MDw can be excluded (see ESI†). For a more straightforward procedure we developed ECC-MW estimation software, in which all current and up-coming ECC-MW estimation techniques and calibration curves as well as different references can be selected and applied. With this software estimated MWdet in THF were compared to MWs of likely monomeric [CpM·THFx] and dimeric aggregates [(CpM)2·THFx] with x = 0–4, M = Li, Na, K, Rb, Cs (see Table 1 or ESI†). The results fit best for monomeric aggregates with different quantities of coordinated THF per alkali metal (except for CpCs, see Table 1 and Scheme 2), hence we propose this to be the most populated species for CpMs in THF solution. Just considering the MWdet of dimeric aggregates, degraded by most coordinated THF they would also be an option (see ESI†). Metallocene-like aggregates of e.g. [Cp2M·THFx]− were not considered because they could not be differentiated from [CpM·THFx+1], because MW(THF) ≈ MW(Cp).
| Monomer + 2 THF | Monomer + 3 THF | Dimer + 4 THF | ||
|---|---|---|---|---|
| MWdet [g mol−1] | MWdif [%] | MWdif [%] | MWdif [%] | |
| CpLi | 218(11) | −1 | 32 | 65 |
| CpNa | 295(16) | −21 | 3 | 27 |
| CpK | 335(18) | −26 | −4 | 17 |
| CpRb | 294(16) | 0 | 25 | 49 |
| CpCs | 1855(198) | — | — | — |
 | ||
| Scheme 2 [CpM] molecules in THF solution. | ||
For CpLi the predominant monomeric aggregate seems to be coordinated by only two THF molecules. This molecule is stable over a large temperature range. If normalised logDx,norm are compared, there is almost a perfect fit for all temperatures: logDx,norm(CpLi, +50) = −8.886; logDx,norm(CpLi, +25 °C) = −8.901; logDx,norm(CpLi, −50 °C) = −8.905; logDx,norm(CpLi, −80 °C) = −8.909; ΔlogDx,norm(CpLi, +50/−80 °C) = 0.023.
The formation of CIPs for CpLi in THF can also be confirmed. This behaviour can be deduced from the same logDx,norm values from the 7Li- and 1H-DOSY NMR spectra (see Fig. 1 and ESI†). Unfortunately, no signal was observable in the 7Li–1H-HOESY NMR experiment to confirm this. For CpNa and CpK coordination by 3 THF molecules is preferred with a slightly bigger MWdif for CpK. After cooling CpNa to −50 °C the same result could be obtained (logDx,norm(CpNa, 25 °C) = −8.978); logDx,norm(CpNa, −50 °C) = −8.971; ΔlogDx,norm (CpNa, +25/+50 °C) = 0.007. It was previously proposed that the mono-solvated [CpNa·THF] is a possible aggregate in the solid state, which we could not confirm in solution.12 Cooling led to precipitation of CpK, CpRb and CpCs, therefore no further insight could be gained for thermal dependence. CpRb seems to be coordinated by only 2 THF molecules at 25 °C. In the literature a coordination polymer of CpRb chains is known, which crystallised with two THF molecules attached to the metal. This provides plausibility to the deaggregation in solution by breaking one of the two Cp–Rb bonds.39 Supposedly this is due to the fading donor capacity of THF descending Group 1. This was e.g. calculated for alkali metal/ammonia binding energies (ΔE(M+–NH3); 170.3 (Li), 117.2 (Na), 82.0 (K), 71.1 (Rb), 61.9 (Cs) kJ mol−1).40 We recently observed the same trend in hexamethyldisilazides crystallized from liquid ammonia.41 The same could be valid for ΔE(M+–THF) as the heavier metal Cp compounds tend to form coordination polymers instead of taking on more coordinated solvent molecules. Rb seems to be the borderline case between not adding donor bonds from solvents and giving up the coordination polymer bonds. DOSY NMR spectroscopic measurements showed that CpCs forms oligomeric aggregates with a MW > 1500 g mol−1 (logDx,norm(CpCs, +25 °C) = −9.407), which also stayed intact after heating to +50 °C (logDx,norm(CpCs, +50 °C) = −9.409, ΔlogDx,norm (CpCs, +25 °C/+50 °C) = 0.002), and it seems that a very specific aggregate is formed because coordination polymer bonds are more important to caesium than to rubidium. In 1996 Harder and Prosenc could show the formation of a caesocene-tripledecker.17 From this and the estimated MW, we envisage a pentameric [(CpCs)5·THF10] (MWdif(merge) = −8%) or hexameric [(CpCs)6·THF12] cyclic structure (MWdif(merge) = 12%),42 whereas other motifs with different amounts of THF or Cp are feasible. Still, it needs to be emphasised that current ECCs are not optimised for aggregates that are that heavy since reference compounds do not cover MWs > 600 g mol−1 yet.
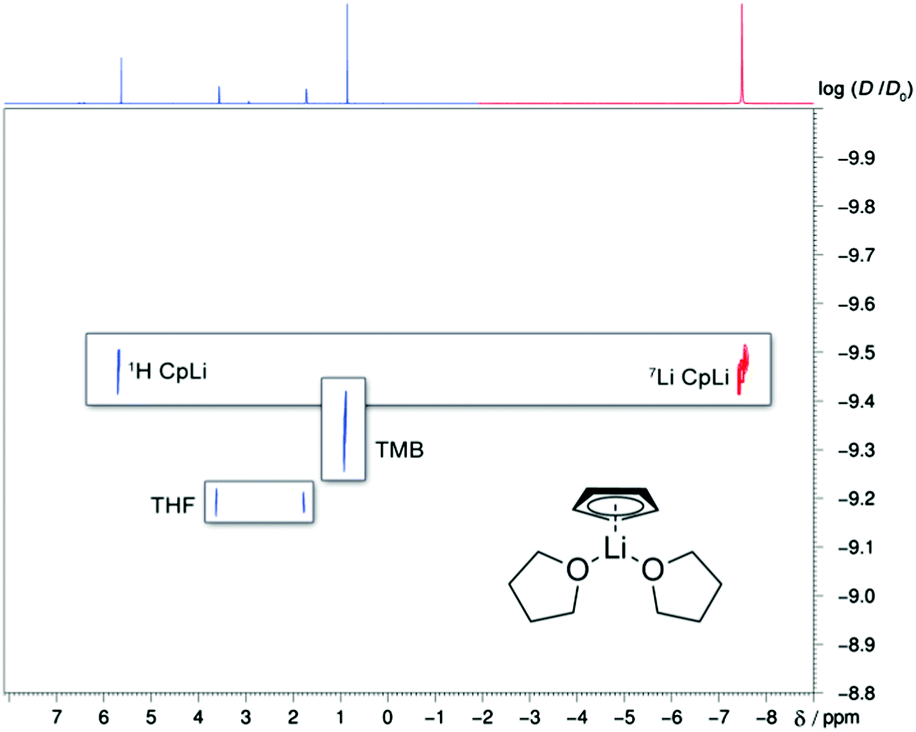 | ||
| Fig. 1 Superposition of 1H- (blue) and 7Li-DOSY (red) NMR spectra of CpLi in THF-d8 at −50 °C. Since only 15 mm solutions of CpLi were used, not all THF is coordinated to CpLi and the diffusion coefficient is averaged with “free” THF. | ||
Furthermore, we measured a sample of CpLi in ammonia solution (see ESI†), where the formation of SSIPs was expected, and saw a change in chemical shift for the 7Li NMR spectroscopic signal towards −0.59 ppm. Moreover, a coupling between ammonia and lithium in a 7Li–1H-HOESY experiment could be observed, which confirms the formation of SSIPs.
In conclusion, with the new ECC-DOSY NMR method we could analyse the donor solvent coordination in alkali metal cyclopentadienides. In THF solution they form monomers and quite surprisingly the lithium and rubidium derivatives only coordinate two solvent molecules while sodium and potassium accommodate three. Presumably the caesium derivative forms a pentamer with two THF molecules coordinated to each metal. We found SSIPs for CpLi in ammonia solution. In addition, we introduced our new ECC-MW estimation software available for anyone to use from our website.
All NMR experiments were recorded on either a Bruker Avance 400 spectrometer equipped with an observer broadband probe with z-axis gradient coil with maximum gradient strength of 57 G cm−1 or Bruker Ascend 400 spectrometer equipped with an inverse broadband probe with z-axis gradient coil with maximum gradient strength of 51 G cm−1. All spectra were acquired in 5 mm NMR tubes. Sample spinning was deactivated during measurements. All DOSY spectra were recorded using the standard Bruker dstebpgp3s pulse sequence with three spoil gradients with convection compensation.43,44 The diffusion time was Δ = 0.1 s. The duration of the magnetic field pulse gradients δ/2 was adjusted for every compound in a range of 1–3 ms (2 to 7 ms for 7Li). The delay for gradient recovery was 0.2 ms and the eddy current delay 5 ms. For each DOSY-NMR experiment, a series of 16 spectra on 32 K data points was collected. The pulse gradients were incremented from 2 to 98% of the maximum gradient strength in a linear ramp with a total experiment time of 51 min. The temperature was set and controlled at 298 K with an air flow of 400 l h−1 in order to avoid any temperature fluctuations due to sample heating during the magnetic field pulse gradients if not stated otherwise. After Fourier transformation and baseline correction, the diffusion dimension was processed with the Topspin 3.1 software. Diffusion coefficients were calculated by exponential fits with the T1/T2 software of Topspin. All samples have been prepared inside a glove box. THF-d8 is stored over 4 Å molecular sieves under argon. All samples were prepared using 15 mM solutions of analyte and internal reference (2,2,3,3-tetramethylbutane (TMB) or 1,2,3,4-tetraphenylnaphthalene (TPhN)). For NMR measurements in NH3, gaseous NH3 was introduced into the NMR tube for one minute at −78 °C; afterwards 0.1 mL of toluene-d8 was added for referencing and after sealing the NMR tube spectra were recorded at ambient temperature. Diffusion coefficients of compounds in THF-d8 were normalised either with the fixed TPhN signal of logDref,fix(TPhN) = −9.1054 or the fixed TMB signal of logDref,fix(TMB) = −8.7749.
We are grateful to the DNRF funded Center of Materials Crystallography (DNRF93) and we appreciate chemical donations from Rockwood Lithium.
Notes and references
- N. J. Long, Metallocenes: An Introduction to Sandwich Complexes, Wiley-Blackwell, 1997 Search PubMed.
- S. Harder, Coord. Chem. Rev., 1998, 176, 17–66 CrossRef CAS.
- A. Raith, P. Altmann, M. Cokoja, W. A. Herrmann and F. E. Kühn, Coord. Chem. Rev., 2010, 254, 608–634 CrossRef CAS.
- T. Stey and D. Stalke, Lead structures in lithium organic chemistry in The chemistry of organolithium compounds, ed., Z. Rappoport and I. Marek, John Wiley & Sons, New York, 2004, pp. 47–120 Search PubMed.
- J. Thiele, Ber. D. Chem. Ges., 1900, 33, 666–673 CrossRef.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- D. Stalke, Angew. Chem., 1994, 106, 2256–2259 ( Angew. Chem., Int. Ed. , 1994 , 33 , 2168–2171 ) CrossRef CAS.
- P. Jutzi and N. Burford, Chem. Rev., 1999, 99, 969–990 CrossRef CAS PubMed.
- R. E. Dinnebier, U. Behrens and F. Olbrich, Organometallics, 1997, 16, 3855–3858 CrossRef CAS.
- R. E. Dinnebier, F. Olbrich, S. VanSmaalen and P. W. Stephens, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 153–158 CrossRef.
- R. E. Dinnebier, F. Olbrich and G. M. Bendele, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 699–701 Search PubMed.
- C. M. Widdifield, J. A. Tang, C. L. B. Macdonald and R. W. Schurko, Magn. Reson. Chem., 2007, 45, S116–S128 CrossRef CAS PubMed.
- R. Michel, R. Herbst-Irmer and D. Stalke, Organometallics, 2010, 29, 6169–6171 CrossRef CAS.
- R. Michel, R. Herbst-Irmer and D. Stalke, Organometallics, 2011, 30, 4379–4386 CrossRef CAS.
- J. Hey, D. M. Andrada, R. Michel, R. A. Mata and D. Stalke, Angew. Chem., 2013, 125, 10555–10559 ( Angew. Chem., Int. Ed. , 2013 , 52 , 10365–10369 ) CrossRef.
- S. Harder and M. H. Prosenc, Angew. Chem., 1994, 106, 1830–1832 ( Angew. Chem., Int. Ed. , 1994 , 33 , 1744–1746 ) CrossRef CAS.
- S. Harder and M. H. Prosenc, Angew. Chem., 1996, 108, 101–103 ( Angew. Chem., Int. Ed. , 1996 , 35 , 97–99 ) CrossRef.
- T. Kaehler and F. Olbrich, Private Communication, CCDC 185158, 2002.
- K. F. Morris and C. S. Johnson, J. Am. Chem. Soc., 1992, 114, 3139–3141 CrossRef CAS.
- D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2009, 42, 270–280 CrossRef CAS PubMed.
- A. Gierer and K. Wirtz, Z. Naturforsch., A: Phys. Sci., 1953, 8, 532 Search PubMed.
- H. C. Chen and S. H. Chen, J. Phys. Chem., 1984, 88, 5118–5121 CrossRef CAS.
- C. A. Crutchfield and D. J. Harris, J. Magn. Reson., 2007, 185, 179–182 CrossRef CAS PubMed.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., 2013, 125, 3281–3284 ( Angew. Chem., Int. Ed. , 2013 , 52 , 3199–3202 ) CrossRef.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- R. Neufeld, M. John and D. Stalke, Angew. Chem., 2015, 127, 7100–7104 ( Angew. Chem., Int. Ed. , 2015 , 54 , 6994–6998 ) CrossRef.
- R. Neufeld, T. L. Teuteberg, R. Herbst-Irmer, R. A. Mata and D. Stalke, J. Am. Chem. Soc., 2016, 138, 4796–4806 CrossRef CAS PubMed.
- D. Stalke and R. Neufeld, Chem. – Eur. J., 2016, 22, 12624–12628 CrossRef PubMed.
- C. Schnegelsberg, S. Bachmann, M. Kolter, T. Auth, M. John, D. Stalke and K. Koszinowski, Chem. – Eur. J., 2016, 22, 7752–7762 CrossRef CAS PubMed.
- S. Bachmann, R. Neufeld, M. Dzemski and D. Stalke, Chem. – Eur. J., 2016, 22, 8462–8465 CrossRef CAS PubMed.
- The fixed diffusion coefficient logDref,fix of TPhN or TMB was estimated by using the middle logD value of multiple DOSY measurements of 15 mM solutions at 25 °C.
- MWdif = [(MWcalc − MWdet)/MWdet] × 100%, where MWdet is the ECC-determined MW of the analyte and MWcalc the calculated MW.
- R. H. Cox and H. W. Terry Jr, J. Magn. Reson., 1974, 14, 317–322 CAS.
- E. Herdtweck, F. H. Köhler and R. Mölle, Eur. J. Inorg. Chem., 2005, 952–958 CrossRef CAS.
- L. A. Paquette, W. Bauer, M. R. Sivik, M. Bühl, M. Feigel and P. V. R. Schleyer, J. Am. Chem. Soc., 1990, 112, 8776–8789 CrossRef CAS.
- J. Wessel, E. Lork and R. Mews, Angew. Chem., 1995, 107, 2565–2567 ( Angew. Chem., Int. Ed. , 1995 , 34 , 2376–2378 ) CrossRef.
- A term coined “molar van-der-Waals density” can be calculated with MDW = MW/∑VW, where ∑VW is the sum of the van-der-Waals volumina of all atoms of a specific molecule. This MDW can be used to account for errors in ECC-MW-calculation, when using compounds incorporating heavier elements. For detailed informations see ESI†.
- U. Behrens and F. Olbrich, Private Communication, CCDC 687263, 2008.
- M. Kaupp and P. V. R. Schleyer, J. Phys. Chem., 1992, 96, 7316–7323 CrossRef CAS.
- R. Neufeld and D. Stalke, Chem. – Eur. J., 2016, 22, 12340–12346 CrossRef CAS PubMed.
- Multihapto bonding is known to prevail with larger and less charge-localising cations like Cs+; see: (a) D. Hoffmann, W. Bauer, P. V. R. Schleyer, U. Pieper and D. Stalke, Organometallics, 1993, 12, 1193–1200 CrossRef CAS; (b) U. Pieper and D. Stalke, Organometallics, 1993, 12, 1201–1206 CrossRef CAS.
- A. Jerschow and N. Müller, J. Magn. Reson., 1996, 222–225 CrossRef.
- A. Jerschow and N. Müller, J. Magn. Reson., 1997, 372–375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Includes detailed information about the DOSY measurements, preparation, ECC-MW estimation and NMR assignments. ECC-MW estimation software can be downloaded at: http://www.stalke.chemie.uni-goettingen.de/mwestimation/. See DOI: 10.1039/c6cc07273a |
| This journal is © The Royal Society of Chemistry 2016 |