
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving O2 reduction at an enzymatic biocathode: mimicking the lungs†
David P.
Hickey
,
Krysti L.
Knoche
,
Kelan
Albertson
,
Carolina
Castro
,
Ross D.
Milton
and
Shelley D.
Minteer
*
Departments of Chemistry and Materials Science and Engineering, University of Utah, Salt Lake City, Utah 84112, USA. E-mail: minteer@chem.utah.edu
First published on 20th October 2016
Abstract
Here, we demonstrate the use of phospholipid micelles to enhance O2 concentrations by two-fold at the surface of a bilirubin oxidase biocathode. Specifically, 1,2-diarachidoyl-sn-glycero-3-phosphocholine was used in a glucose enzymatic fuel cell to limit power losses due to O2 transport, even in a quiescent solution.
Enzymatic fuel cells (EFCs) have rapidly developed as a possible power source for small implantable electronic devices. Utilizing an enzymatic catalyst at one or both the anode and cathode, EFCs are capable of operating under physiological pH and temperature. Advancements in enzymatic anode materials have provided dramatic improvements in bioanodic current density; however, comparatively little progress has been made towards biocathode development despite being commonly cited as limiting to the current output of EFCs.1–6 Unlike bioanodic substrates such as saccharides and short-chain alcohols that are highly water-soluble, biocathodic substrates are almost exclusively limited to reducible gases, such as molecular oxygen, that are only sparsely soluble in aqueous solutions.7 Organic solvents, such as hexane and perfluorodecalin, are highly effective at solubilizing O2, but are incompatible with most biological systems.8,9 Hence, further biocathode development requires broadly applicable strategies for increasing O2 concentration at the enzyme-modified electrode interface. Consequently, we considered utilizing lipid micelle solutions as a way to combine the high-oxygen solubility of organic solvents with the biocompatibility of aqueous solutions. Herein, we describe the ability of a phospholipid, phosphatidylcholine, to increase the apparent O2 concentration at a bilirubin oxidase (BOx) biocathode, allowing for dramatically improved quiescent biocathodic current density.
Enzymatic biocathodes are generally constructed using a polymer to immobilize an O2-reducing enzyme, such as laccase or bilirubin oxidase, onto the surface of a carbon electrode. During immobilization, the enzyme is either combined with a redox mediator to act as an electron shuttle between the electrode and the enzyme's active site,10–12 or the enzyme is oriented toward the electrode surface to facilitate efficient direct electron transfer (DET).11,13–15 An extensive body of literature has demonstrated methods for enhancing electron transfer rates between O2-reducing enzymes and electrode surfaces through either mediator-assisted electron transfer or DET; however, the maximum current densities (jmax) for these systems can only be reached through a combination of rapid convection and O2-bubbling throughout the electrochemical measurements. This highlights the need for techniques to improve O2 transport, and indicates that further biocathode development requires that these challenges be met.
Calabrese Barton and co-workers predict that biocathodes should theoretically be able to achieve current densities of 60 mA cm−2, wherein they list the primary obstacles to an ideal biocathode as enzyme surface coverage and O2 transport.16 Even if enzyme surface coverage and electron transfer can be made ideal, the necessity of rapid convection and O2-bubbling adds a substantial energy cost to fuel cell operation and limits the practical application of the resulting EFCs. Therefore, successful strategies for increasing O2 solubility in a biocathode solution must specifically aim to increase the local transport of O2 to the biocathode surface.
Artificial oxygen carriers (AOCs) have long been studied to address medical problems associated with hypoxia. The most widely studied AOCs for in vivo use include O2-saturated hemoglobin derivatives and perfluorocarbon formulations; however, the application of these solutions in the context of EFCs is complicated by slow intermolecular O2 transport and biological incompatibility as mentioned before.17,18 A possible alternative solution to the problem of aqueous O2 solubility can be found in the human respiratory system, which uses phosphatidylcholine derivatives to enhance O2 transport into the blood stream.19 Phospholipids make up the primary surfactant class found in the lungs to solubilize O2 from the air into the blood stream. In this process, O2 is solubilized into phospholipid surfactant bilayers upon expansion of lungs and subsequently absorbed into alveolar cells where it is transferred into the blood stream. Pulmonary surfactants exist primarily in the solid phase, through which O2 diffuses very slowly.20 It is only the expansion of the lungs that creates the increased partial pressure necessary to force O2 into the lipid bilayer. Based on this, we hypothesized that a solid phase phospholipid surfactant could be used to trap and store O2 in an aqueous solution, thereby enhancing the effective concentration at a biocathode interface.
To initiate our investigation, we screened a range of commercially available phospholipids to determine their capacity to trap and store O2. This was accomplished amperometrically by bubbling O2 through a phospholipid solution and, using a platinum rotating disk electrode (RDE) as an oxygen reduction catalyst, measuring the amount of time (t1/2) required to reach 50% of jmax (detailed procedures and complete screening results provided in ESI,† Table S1 and Fig. S2). An initial screen was performed on short chain phosphocholines such as 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), L-α-phosphatidylcholine (L-α-PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (DPTE) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG)2000] (DSPE-PEG2000). Solutions of L-α-PC, DOPE, DPTE and DSPE-PEG2000 all exhibited t1/2's similar to solutions containing only buffer (400–500 s compared to 460 s in buffer). However, DSPC exhibited a residence time (880 ± 90 s) nearly double that of solutions containing buffer alone.
Based on our initial hypothesis, we suspected that the anomalously high retention time of DSPC would be coupled to its state of phase under the temperature being studied. To probe this supposition, we expanded our initial screen to include phosphocholines of varying chain length, and thus varying transition temperatures (Fig. 1). It should be noted that throughout the remainder of the manuscript, phosphocholines (PCs) of varying length will be referred to by their chain length for systematic simplicity (i.e.PC12, PC14, PC16, PC18 and PC20 refer to DLPC, DMPC, DPPC, DSPC and DAPC, respectively). A comparison of t1/2 with the corresponding transition temperature of the lipid reveals that PCs with a transition temperature above the experimental temperature exhibit significantly longer retention times; this supports the proposed correlation between state of phase and t1/2. While RDE experiments were able to shed light on the nature of O2 retention time for solutions of various phospholipids, the low catalytic activity of Pt under neutral pH and ambient temperature resulted in minimal variation in jmax.
 | ||
| Fig. 1 PC phase transition temperature versus retention time (t1/2) for solutions containing 0.5 mM PC. Retention time was measured amperometrically by platinum disk electrodes rotated at 500 rpm using 200 mM citrate/phosphate buffer at pH 6.5 and 25 °C. | ||
With the primary goal of this work aimed at utilization in an EFC, we next sought to determine the impact of PCs on apparent O2 concentration and retention time using a previously reported biocathode design based on bilirubin oxidase (BOx) with anthracene-modified carbon nanotubes on a 1 cm2 Toray carbon paper electrode.21 Cyclic voltammetry (CV) was performed using BOx biocathodes suspended in O2-saturated buffers containing PCs of various chain length (Fig. 2). The resulting data indicates that the presence of PC with any length of chain increases jmax of the biocathode (ranging from jmax = 0.73 ± 0.20 mA cm−2 in PC12 to jmax = 1.14 ± 0.15 mA cm−2 in PC20). However, similar to the RDE experiments, there is no statistical difference between jmax obtained using PC12, PC14 or PC16, while the apparent O2 concentration is increased in the presence of either PC18 or PC20 (jmax = 0.94 ± 0.14 mA cm−2 and jmax = 1.14 ± 0.15 mA cm−2, respectively). A more thorough analysis of CV results and experimental procedures is provided in the ESI† (Fig. S3–S5).
 | ||
Fig. 2 Representative CVs of BOx biocathodes under N2 saturation (—), quiescent O2 saturation ( ) or stirred O2 saturation ( ) or stirred O2 saturation ( ) in solutions containing buffer alone or with 0.5 mM PC12, PC14, PC16, PC18 or PC20. Experiments were performed at 5 mV s−1 using 200 mM citrate/phosphate buffer, pH 6.5, at 25 °C. ) in solutions containing buffer alone or with 0.5 mM PC12, PC14, PC16, PC18 or PC20. Experiments were performed at 5 mV s−1 using 200 mM citrate/phosphate buffer, pH 6.5, at 25 °C. | ||
In order to provide a distinction between O2 transport and localized O2 concentration, jmax was measured under both stirred and quiescent conditions. The resulting CVs reveal that, as expected, jmax increases with stirring for PC12–18; this reflects increased O2 transport to the electrode surface. However, the difference in stirred vs. quiescent jmax decreases as PC chain length increases to the extent that stirring lowers jmax in solutions of PC20. While the precise nature of this result is unclear, it indicates that the phospholipids are allowing for increased local O2 concentration at the surface of the electrode as opposed to increasing O2 concentration throughout the bulk solution.
We next aimed to determine the duration of these unexpected effects. To accomplish this, O2 retention times at BOx biocathodes were measured amperometrically as above using stirred solutions containing PCs of various length (Fig. 3). The resulting t1/2 values demonstrate a similar correlation between state of phase and retention time that was observed at Pt RDEs. However, O2 retention time measured by BOx biocathodes was greater than 10 times longer for solutions of PC20 (1800 ± 200 s) than for buffer (270 ± 100 s). Additionally, the increase in t1/2 for all studied PCs beyond the control buffer was significantly greater when studied at the BOx biocathodes than the Pt RDE, suggesting a fundamental difference in the mechanism of O2 transport between the biocathode and Pt electrode interface.
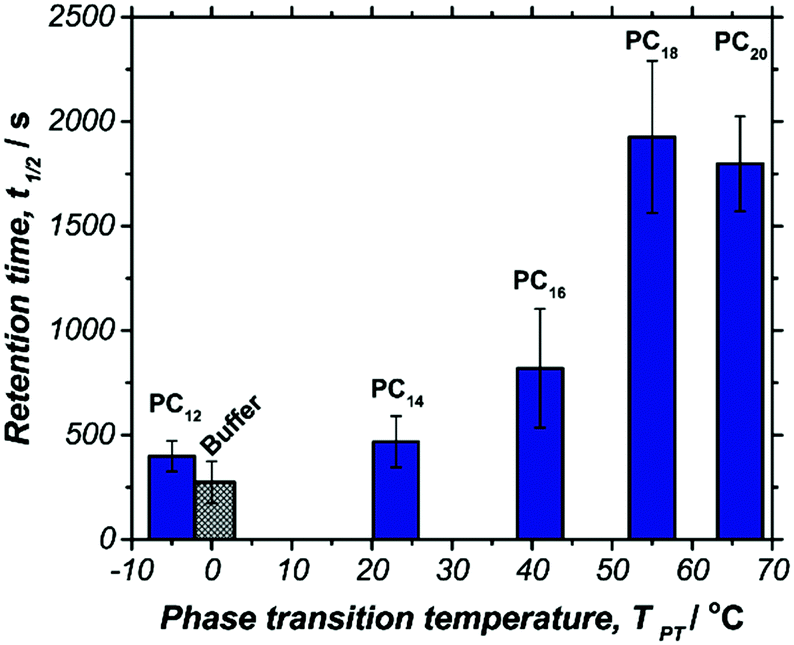 | ||
| Fig. 3 PC phase transition temperature versus retention time (t1/2) for solutions containing 0.5 mM PC. Retention time was measured amperometrically by 1 cm2 BOx biocathodes using 200 mM citrate/phosphate buffer at pH 6.5 and 25 °C. | ||
Based on the large discrepancy in t1/2 between electrode materials, we turned our attention towards methods to study the BOx biocathode surface in the presence of PCs. Unfortunately, fluorescence images of BOx-modified Toray paper electrodes revealed a strong background fluorescence from the anthracene-modified CNTs used to prepare the electrode material. Consequently, a (7-nitro-2-1,3-benzoxadiazol-4-yl)amino (NBD)-modified PC18 was used as a fluorescent probe to compare the behavior of PCs in solution with that at the surface of an unmodified Toray electrode. Fluorescence images of NBD-PC18 (provided in the ESI,† Fig. S9–S28) indicate that solid-phase lipids in solution are distributed homogeneously, while similar images of NBD-PC18 in the presence of a Toray paper electrode reveal the formation of phospholipid aggregates on the electrode surface. Taken together with the observed increase in catalytic current density of BOx biocathodes in quiescent solutions, this suggests that PC18 and PC20 aggregates are trapping O2 at the biocathode surface, allowing for enhanced localized O2 concentration. (Full fluorescence analysis of phospholipid interactions at the biocathode surface are provided in the ESI†).
Having established the use of PC20 to enhance the apparent O2 concentration at a BOx biocathode surface, the next step was to determine its compatibility in a fully enzymatic biofuel cell. We employed a recently reported glucose bioanode that uses a naphthoquinone-modified linear poly(ethylenimine) (NQ-LPEI) hydrogel to immobilize flavin adenine dinucleotide-dependent glucose dehydrogenase (FAD-GDH).4 This material utilizes naphthoquinone as a low-potential electron mediator to allow for increased cell potential, while the FAD-GDH does not use O2 as a natural electron acceptor and as such, mediated enzymatic glucose oxidation does not compete with enzymatic O2 reduction.
EFCs were constructed to have a bioanode and biocathode of equal surface area (1 cm × 1 cm Toray electrodes) and were characterized by linear polarization at 1 mV s−1 using solutions of 100 mM glucose. Power and current density curves of EFCs tested using quiescent solutions in the presence and absence of 1 mM PC20 are presented in Fig. 4. EFCs performed using solutions with 1 mM PC20 exhibited a 10-fold increase in current density (jmax) and power density (Pmax) compared to those tested in quiescent solutions containing glucose alone (jmax = 1.9 ± 0.4 mA cm−2 and Pmax = 0.8 ± 0.2 mW cm−2versus jmax = 0.100 ± 0.009 mA cm−2 and Pmax = 0.061 ± 0.005 mW cm−2). Comparison of the corresponding power curves indicates that the majority of the increase in current density occurs at low resistance in the diffusion-limited region of the curve. Combined with control experiments (provided in the ESI†) demonstrating a high limiting anodic current density (jmax = 2.2 mA cm−2), these results suggest that the increased jmax and Pmax are a result of increased O2 concentration at the biocathode.
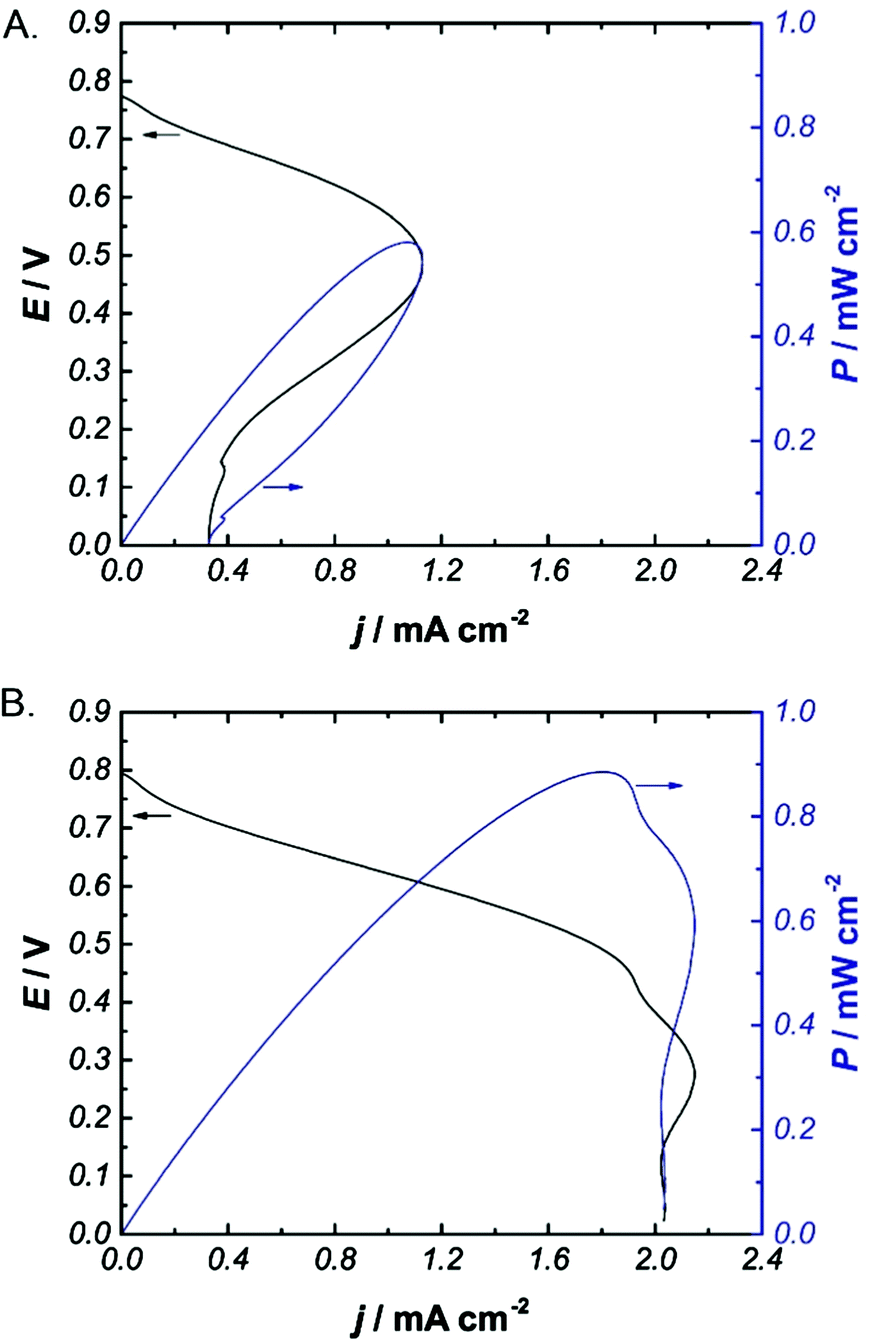 | ||
| Fig. 4 Representative power and current density curves for EFCs in a quiescent solution without (A) or with (B) 1 mM PC20. EFCs were constructed with naphthoquinone-modified LPEI (NQ-LPEI)/FAD-GDH bioanodes and anthracene-modified carbon nanotubes (An-CNTs)/BOx using solutions of 200 mM citrate phosphate buffer, pH 6.5, with 100 mM glucose at 25 °C. | ||
In conclusion, we have identified a series of commercially available phospholipids capable of trapping O2 in an aqueous solution and increasing its residence time at the surface of a biocathode. Additionally, the solid-phase phosphocholine, PC20, was used to increase the local apparent O2 concentration at the surface of a BOx-modified biocathode, resulting in a catalytic current density of 1.1 ± 0.1 mA cm−2 for ORR at 25 °C and physiological pH. While this methodology was described in the context of EFCs, it has a direct potential application in ex vivo microbial fuel cells. Furthermore, we believe that this work will provide a foundation for engineering solutions to address the problem of aqueous O2 transport. Finally, PC20 was demonstrated as a simple EFC additive to enhance current and power outputs without the need for artificial convection.
The authors would like to thank the National Science Foundation, Air Force Office of Scientific Research, and the Army Research Office for funding. The authors would also like to thank Matt Judge and Derek Jensen for collecting the EFC data.
Notes and references
- W. Schuhmann, T. J. Ohara, H. L. Schmidt and A. Heller, J. Am. Chem. Soc., 1991, 113, 1394–1397 CrossRef CAS.
- M. T. Meredith, D.-Y. Kao, D. Hickey, D. W. Schmidtke and D. T. Glatzhofer, J. Electrochem. Soc., 2011, 158, B166–B174 CrossRef CAS.
- B. Reuillard, A. Le Goff, C. Agnes, M. Holzinger, A. Zebda, C. Gondran, K. Elouarzaki and S. Cosnier, Phys. Chem. Chem. Phys., 2013, 15, 4892–4896 RSC.
- R. D. Milton, D. P. Hickey, S. Abdellaoui, K. Lim, F. Wu, B. Tan and S. D. Minteer, Chem. Sci., 2015, 6, 4867–4875 RSC.
- S. Tsujimura, Y. Kamitaka and K. Kano, Fuel Cells, 2007, 7, 463–469 CrossRef CAS.
- K. MacVittie, T. Conlon and E. Katz, Bioelectrochemistry, 2015, 106(Part A), 28–33 CrossRef CAS PubMed.
- R. Battino, T. R. Rettich and T. Tominaga, J. Phys. Chem. Ref. Data, 1983, 12, 163–178 CrossRef CAS.
- E. P. Wesseler, R. Iltis and L. C. Clark Jr, J. Fluorine Chem., 1977, 9, 137–146 CrossRef CAS.
- E. Katz, B. Filanovsky and I. Willner, New J. Chem., 1999, 23, 481–487 RSC.
- S. Reiter, K. Habermüller and W. Schuhmann, Sens. Actuators, B, 2001, 79, 150–156 CrossRef CAS.
- S. C. Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Phys. Chem. B, 2001, 105, 11917–11921 CrossRef CAS.
- L. Stoica, N. Dimcheva, Y. Ackermann, K. Karnicka, D. A. Guschin, P. J. Kulesza, J. Rogalski, D. Haltrich, R. Ludwig, L. Gorton and W. Schuhmann, Fuel Cells, 2009, 9, 53–62 CrossRef CAS.
- M. Falk, Z. Blum and S. Shleev, Electrochim. Acta, 2012, 82, 191–202 CrossRef CAS.
- K. Stolarczyk, E. Nazaruk, J. Rogalski and R. Bilewicz, Electrochim. Acta, 2008, 53, 3983–3990 CrossRef CAS.
- G. Gupta, V. Rajendran and P. Atanassov, Electroanalysis, 2004, 16, 1182–1185 CrossRef CAS.
- S. Calabrese Barton, Electrochim. Acta, 2005, 50, 2145–2153 CrossRef.
- M. G. Scott, D. F. Kucik, L. T. Goodnough and T. G. Monk, Clin. Chem., 1997, 43, 1724–1731 CAS.
- J. S. Jahr, S. B. Nesargi, K. Lewis and C. Johnson, Am. J. Ther., 2002, 9, 437–443 CrossRef PubMed.
- J. N. Kheir, L. A. Scharp, M. A. Borden, E. J. Swanson, A. Loxley, J. H. Reese, K. J. Black, L. A. Velazquez, L. M. Thomson, B. K. Walsh, K. E. Mullen, D. A. Graham, M. W. Lawlor, C. Brugnara, D. C. Bell and F. X. McGowan, Sci. Transl. Med., 2012, 4, 140ra188 Search PubMed.
- B. Olmeda, L. Villén, A. Cruz, G. Orellana and J. Perez-Gil, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1281–1284 CrossRef CAS PubMed.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683–1690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07215a |
| This journal is © The Royal Society of Chemistry 2016 |