
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blue rhenium tricarbonyl DPPZ complexes – low energy charge-transfer absorption at tissue-penetrating wavelengths†
M. P.
Coogan
 *a and
J. A.
Platts
*b
*a and
J. A.
Platts
*b
aChemistry Department, Lancaster University, Faraday Building, Bailrigg, Lancaster, LA1 4YB, UK. E-mail: m.coogan@lancs.ac.uk
bCardiff School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
First published on 29th September 2016
Abstract
fac-[Re(CO)3(dppz)] complexes of amido ligands generated in situ under basic conditions display an intense (ε = 16460 L mol−1 cm−1) absorption band between 500–700 nm which are assigned to an n(amido,RNH–) → π* (dppz) inter-ligand charge transfer band offering an extraordinarily low energy charge separating absorption with potential for imaging and energy application.
Low energy charge transfer transitions are of great importance in a variety of applications including photovoltaics and other energy related areas. These often involve donor acceptor pairings between materials and small molecules1,2 or polymers3 and in the case of dye-sensitised solar cells a metal-to-ligand charge transfer transition in a ruthenium complex.4 Typically, metal to ligand charge transfer bands of ruthenium bipyridines and related complexes are centred around 400–500 nm ([Ru(bpy)3]2+, H2O, 452 nm) and efficiency in solar energy harvesting requires the development of dyes which absorb lower energy light. Given the vast effort which is currently devoted to developing sensitisers which harvest more of the solar spectrum, any transition which both generates charge separation and can be induced at long wavelengths is of obvious interest.
A dramatic blue colour was observed to immediately form upon addition of ethylene diamine to fac-[Re(CO)3(dppz)(3-chloromethylpyridyl)][BF4] (dppz = dipyrido[3,2-a:2′,3′-c]phenazine) 1 (Fig. 1).5 Further investigation proved this to be a general effect observed upon addition of substituted and unsubstituted aliphatic diamines (ethylene diamine, propylene diamine, 2,2-dimethylpropylenediamine, 1,1-dimethylethylenediamine) but not ammonia, monoamines, tetrasubstituted diamines (TMEDA) or amino alcohols, to all available examples of complexes of the general formula fac-[Re(CO)3(dppz)(L)] (L = Py, halide, OTf or MeCN).
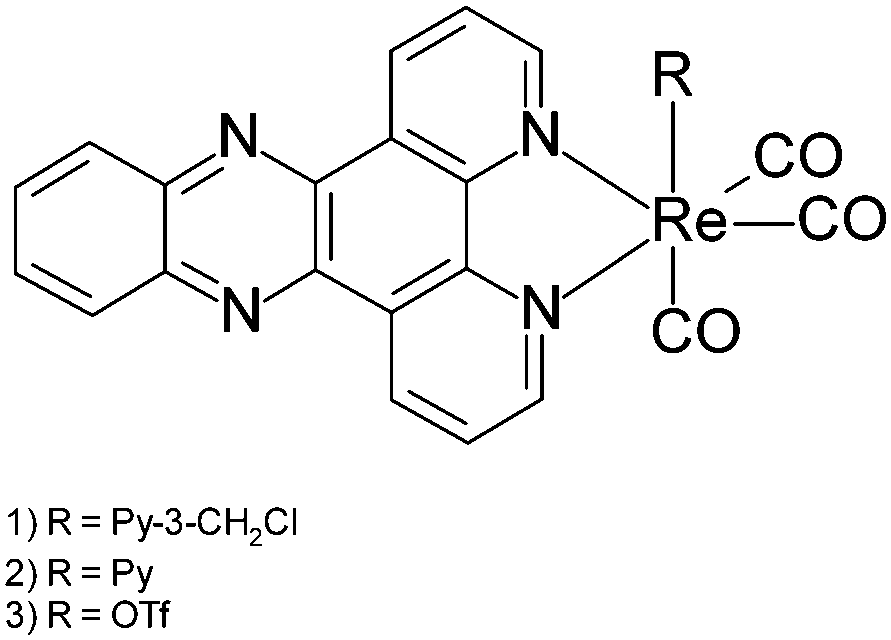 | ||
| Fig. 1 Structures of Re dppz complexes. | ||
The blue colour results from an intense new electronic transition centred at 554 nm, ε = 16460 L mol−1 cm−1 (Fig. 2) and extending into the near IR, which grows in upon the addition of diamines to solutions of complexes. Dppz complexes of a variety of transition metals are known, and they have been widely studies as DNA photoswitches,6–13 but typically their MLCT bands are centred at 400–500 nm giving orange to red complexes with this blue species highly unusual.
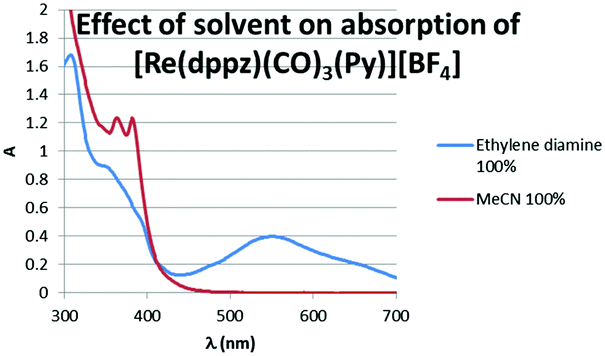 | ||
| Fig. 2 Effect of solvent on absorption spectrum of [Re(dppz)(CO)3(Py)][BF4]. | ||
The IR stretching frequencies for the carbonyls, which are very sensitive to coordination environment show additional bands in the presence of diamine, but with only small shifts indicating exchange of only the ‘axial’ ligand with the [Re(CO)3dppz] core remaining intact (see ESI,† Table S1). Aggregation of complexes induced by the polarity of diamines could lead to species with low energy charge transfer bands, however titrations indicate that the formation of the blue species is dependent upon both diamine concentration and rhenium concentration, with absorption intensity at 560 nm increasing approximately linearly as a function of the concentration of diamine (see ESI,† Fig. S1) and as a function of complex concentration. The absorption at 380 nm (MLCT) varies with addition of diamine (as predicted from the spectra in Fig. 2) but its intensity is not a function of [diamine] but is approximately proportional to [complex]. An aggregation of complexes triggered by the presence of a polar diamine in the solvent mixture should not show such a simple dependency upon both complex and amine concentration, but should be more highly sensitive to the concentration of complex. The new transition appears to result from a transient complex formed upon displacement of the most labile monodentate ligands (Py, halide etc.), as it is reversible, with the low energy band being lost upon dilution of the sample with respect to diamine, with this being most apparent upon addition of protic solvents such as water (see ESI,† Fig. S2).
Given that the phenomenon was not observed with monoamines or tetrasubstituted diamines, it was clear that the second amine was playing an important role in the new band, however it was not clear what: hydrogen bond donation could be precluded by the observation that 1,1-dimethylethylenediamine showed the phenomenon (assuming coordination through the primary amine), and amino alcohols should show hydrogen bond acceptance properties, but did not give blue complexes. It was also noteworthy that the phenomenon was only observed at high concentrations of ethylene diamine, and lower concentrations did not give the same result even with more reactive rhenium starting materials (e.g. [Re(CO)3(dppz)(MeCN)][BF4])5 suggesting that more than one diamine was involved in the production of the blue complex.
NMR spectroscopy (Fig. 3) provided an intriguing insight when a sample of [Re(dppz)(CO)3(Py)][BF4] in a d-3 MeCN/ethylene diamine mixture showed, clearly resolved from the ethylene diamine peaks (<3 ppm), a triplet (∼3.6 ppm, red) and a broad singlet (∼4.2 ppm, green) with relative integration approximating to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 respectively. Additionally the rise in of these new peaks could be correlated with the appearance of free pyridine (blue) and the diminution of the peaks for coordinated pyridine. The triplet was tentatively assigned as a methylene unit of a coordinated ethylene diamine, desymmetrised by coordination and this was confirmed by the approximate 1:1 relative integration of this peak with the peaks assigned to a newly formed complex in the ligand region. The broad singlet was assigned to an NH, arising from coordination of an ethylene diamine amine, followed by deprotonation at higher amine concentrations (i.e. basicity) (Scheme 1 complex 4).
:1 respectively. Additionally the rise in of these new peaks could be correlated with the appearance of free pyridine (blue) and the diminution of the peaks for coordinated pyridine. The triplet was tentatively assigned as a methylene unit of a coordinated ethylene diamine, desymmetrised by coordination and this was confirmed by the approximate 1:1 relative integration of this peak with the peaks assigned to a newly formed complex in the ligand region. The broad singlet was assigned to an NH, arising from coordination of an ethylene diamine amine, followed by deprotonation at higher amine concentrations (i.e. basicity) (Scheme 1 complex 4).
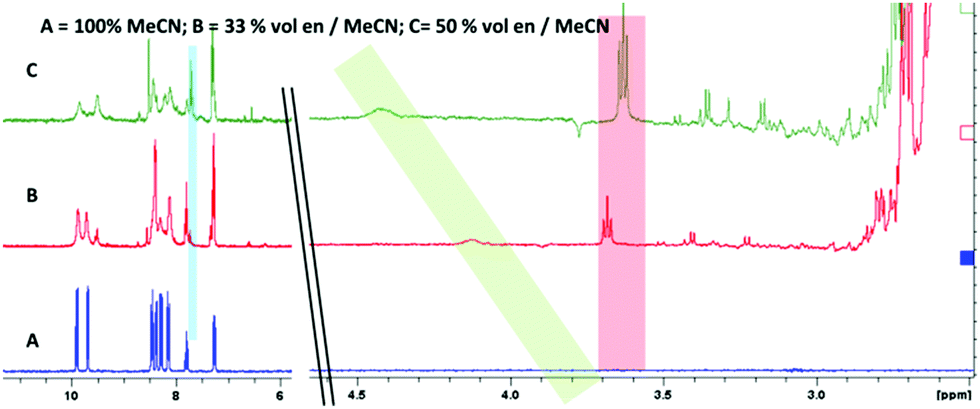 | ||
| Fig. 3 1H NMR of complex 2 in CD3CN with addition of ethylene diamine. Note appearance of triplet from desymmetrised ethylene diamine (highlighted red); broad singlet attributed to amido NH (highlighted green) and free pyridine formed upon displacement (highlighted blue). | ||
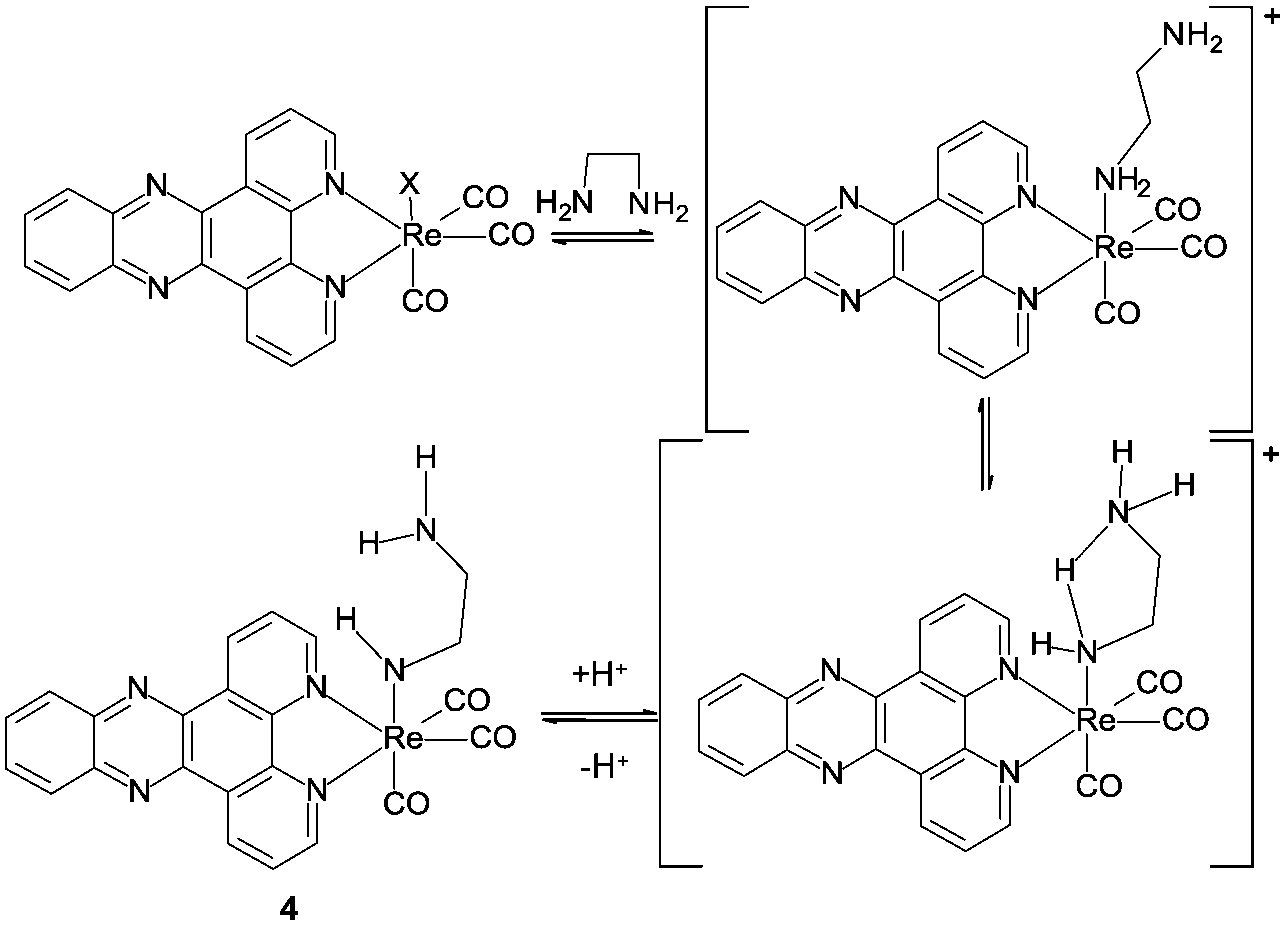 | ||
| Scheme 1 Proposed speciation in ethylene diamine solution. | ||
In order to rationalise this assignment with the observation that the new band is only observed in the presence of diamine and not monoamines, even at concentrations of amine which should give much higher pH values than the minimum diamine concentration under which blue colour appears, a role for the second amine is postulated in assisting proton removal from the coordinated amine through intramolecular hydrogen bonding.
This proposal fitted all the observations made so far: that the phenomenon was only observed with diamines and not monoamines, that it was a function of diamine concentration (related to pH), not stoichiometry with the metal and that 1,1-disubstituted diamines showed the phenomenon, but that tetrasubstituted diamines (TMEDA) did not. However, it did not provide an indication of the electronic nature of this new low energy band and so DFT calculations were undertaken in order to examine its origin. DFT geometry optimisation was performed using the B3LYP functional14,15 with Stuttgart–Dresden basis set and effective core potential for Re,16 along with 6-31+G(d,p) on all non-metal atoms.17–19 Time-dependent DFT calculation of absorption bands at the optimal geometry used the same basis set with the CAM-B3LYP functional,20 and also included a simulated MeCN environment using the polarised continuum model (PCM) approach.21 All calculations were performed in Gaussian09.22
Geometry optimisation supported the proposal of intramolecular hydrogen bonding as an important factor in the structure for complex 4, showing a N–H⋯N distance of 2.599 Å, with the ethylene diamine chain arranged so as to minimise this distance. Examination of the ground state electronic structure by DFT showed that the HOMO is composed of NH lone pair, Re d-orbital Re–CO p-bond trans to ethylene diamine. The LUMO, as well as LUMO+1 and LUMO+2, are almost entirely composed of dppz π* orbitals (Fig. 4).
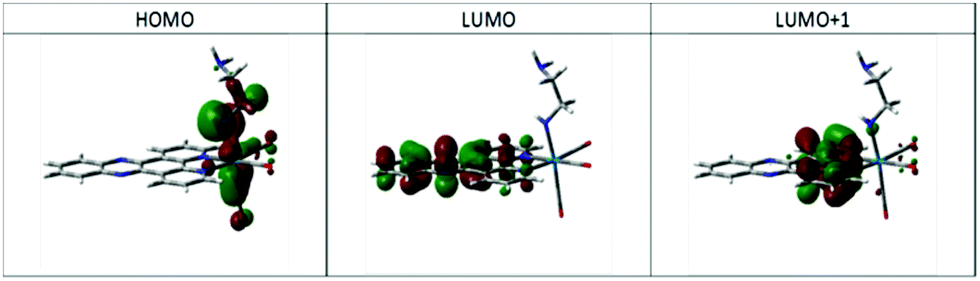 | ||
| Fig. 4 Calculated HOMO and LUMOs of 4. | ||
TD-DFT calculations using the CAM-B3LYP functional in a continuum model of CH3CN solvation predict a low energy transition resulting from HOMO → LUMO/LUMO+1 excitation with wavelength 535 nm, i.e. close to the observed maximum in Fig. 2. The nature of the orbitals involved mean that this can be assigned as n(amido,RNH–) → π*(dppz). Higher energy transitions with greater intensity are found centred on 350 nm, arising from HOMO−1 → LUMO and HOMO−2 → LUMO transitions, which are of essentially MLCT nature (see ESI,† Fig. S3 for orbital plots of HOMO−1 and HOMO−2). Thus it appears that calculations support the assignment of the new low energy absorption band to a deprotonated amido ligand, and furthermore clarify its origin as being of amido lone pair → dppz π* nature (see ESI,† Fig. S3 for CAM-B3LYP predicted spectrum).
In contrast to the low energy n(amido,RNH−) → π* transition offered by a formally anionic amido ligand, TD-DFT suggested that a neutral amine ligand devoid of the uncoordinated lone pair to participate in n(amido,RNH–) → π* transition should show no such low energy band with the lowest energy transition predicted at 329 nm, arising from a combination of HOMO/HOMO−2 → LUMO/LUMO+1 excitation. Orbital plots (see ESI,† Fig. S4) indicate that this transition can be assigned as MLCT in nature.
Experimental support for these findings came when a nearer stoichiometric mix of ethylene diamine and a reactive rhenium precursor, [Re(dppz)(CO)3(OTf)] initially gave only a small change in the spectrum, presumably indicating and equilibrium involving formation of some of the cationic amine complex due to the presence of several equivalents of ethylene diamine, but not at a concentration which is high enough to lead to deprotonation and formation of the amido complex.
However, treatment of this mixture with an external base (NaOtBu) led to immediate appearance of the new band attributed to (amido)n → π* charge transfer (see ESI,† Fig. S5). Given the formation of the new band when an external base is used to deprotonate a coordinated amine it seemed likely that other amines could also show similar bands, and in fact DFT suggested a similar low energy transition would be available for the analogous aniline complex upon deprotonation (Scheme 2). As with the ethylene diamine complex, a low energy transition at 573 nm originates from HOMO (amido lone pair) → to LUMO/LUMO+1 (dppz π*) excitation (see ESI,† Fig. S6 and S7). Experimental support for this was obtained when the same reactive complex [Re(dppz)(CO)3(OTf)] as was used in the external base experiment above was treated with aniline, again giving a small shift in the spectrum, but no low energy band, and, again, upon addition of external base (NaOtBu) the low energy band grew in as a function of basicity (Fig. 5 and 6).
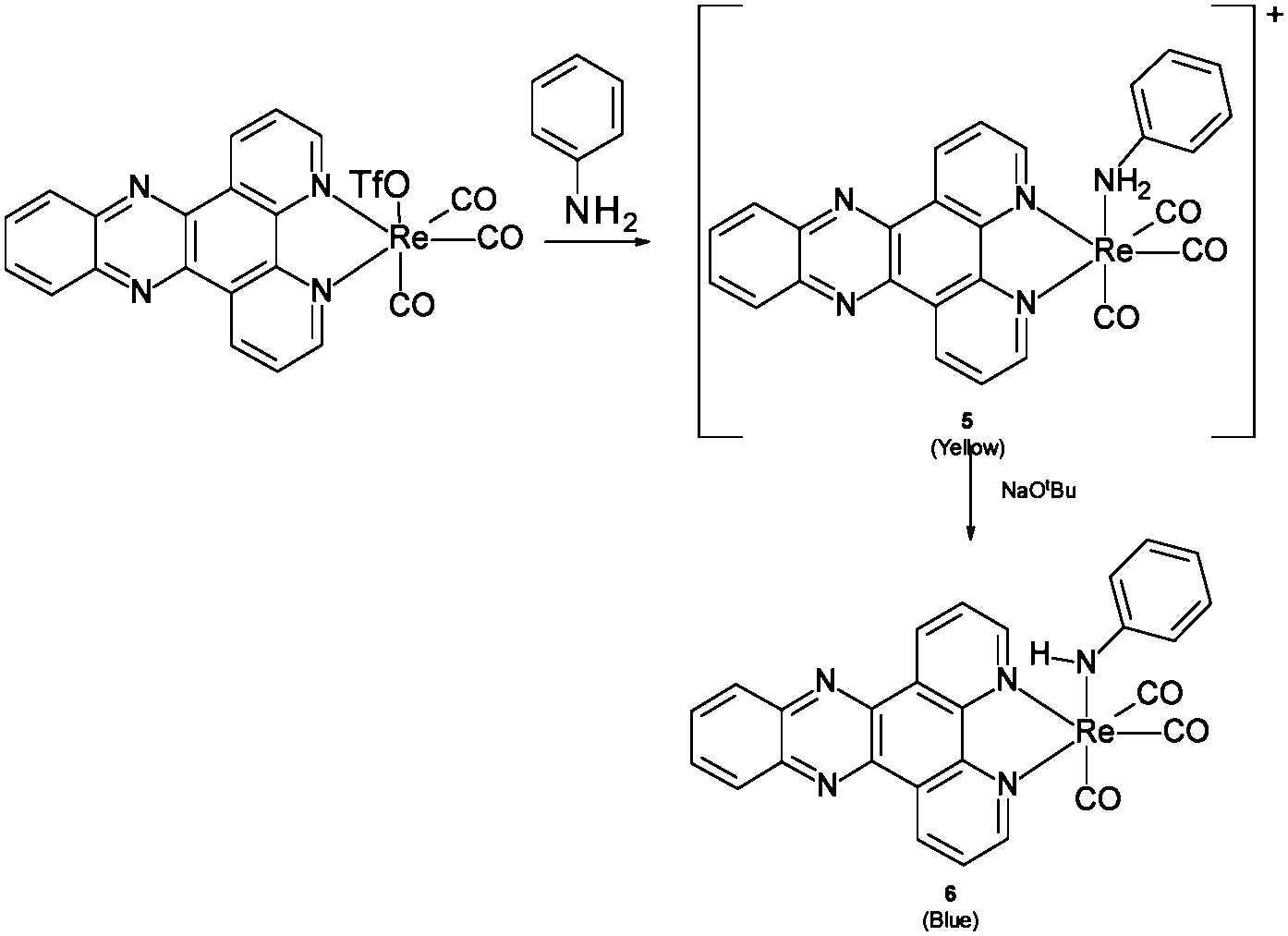 | ||
| Scheme 2 Proposed interaction of 3 with aniline and base. | ||
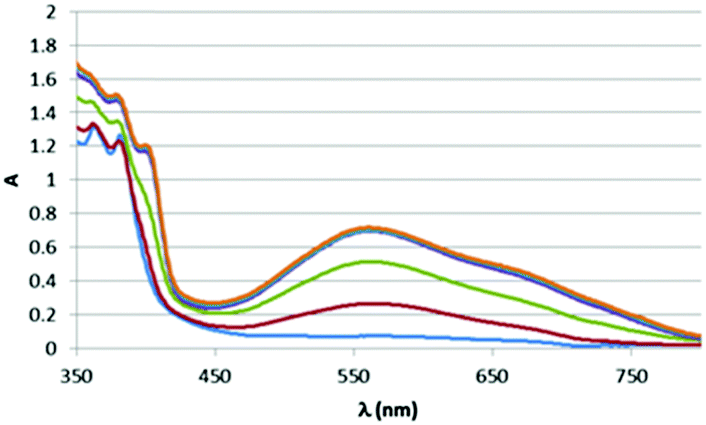 | ||
| Fig. 5 Effect of base on absorption spectrum of stoichiometric mixture of aniline + 1. | ||
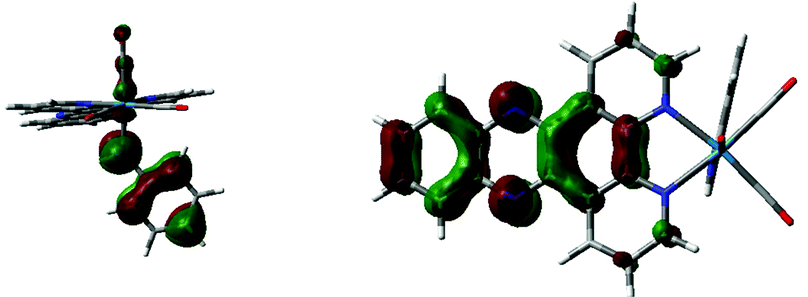 | ||
| Fig. 6 Calculated HOMO (left) and LUMO (right) of aniline amido complex 5. | ||
As much of the interest in metal DPPZ complexes results from their photoluminescence and in particular their emission photoswitching in the presence of DNA, the emission properties of these new amido complexes was examined. Excitation at high energy into the MLCT bands led to visible emission in line with previous studies (λmaxex 345 nm, λmaxem 550 nm),5 however, excitation at the low energy bands associated with the (amido,RNH–) → π* band did not give any detectable luminescence, presumably due to efficient deactivation via non-radiative pathways.
To test the generality of these findings, DFT studies of analogous Mn complexes were carried out. The en-amido Mn complexes is predicted to absorb radiation at 504 nm, i.e. slightly blue-shifted compared to the Re complex discussed above. This again arises from HOMO (amido,RNH–) → LUMO/LUMO+1 (dppz π*) excitation, as shown by the orbital plots below. Similarly, the Mn–aniline complex is predicted to absorb at 533 nm, a slight red shift from the en complex, due to excitation from HOMO to LUMO/LUMO+1 (see ESI,† Fig. S8 and S9). Attempts to reproduce these predictions experimentally were complicated by the labile coordination chemistry of Mn compared to Re. Although low energy bands were apparent upon addition of ethylene diamine, or aniline and base to [MnBr(CO)3(dppz)]‡ there were other changes observed in the spectra indicative of other reactions and more investigation of this underexplored system is required to develop the [Mn(CO)3(dppz)] series.23 Thus we predict on theoretical grounds that this phenomenon is not unique to Re, and that judicious choice of metal and ligand allows fine tuning of the wavelength of maximum absorption.
While Re bipyridine complexes do not show this phenomenon, the proposed orbital origin suggests that it should not be limited to DPPZ complexes, but should be observed with a variety of other highly conjugated diamines which provide a low energy π* orbital.
This observation of the simple formation of red-absorbing complexes is of more academic interest as it represents an unusually low energy charge-separating transition. Such transitions are of interest in a wide range of applications such as e.g. photovoltaics, photodynamic therapy and photocatalysis, and its availability in a range of complexes based on different metals suggests many possible applications. Photodynamic therapy involves activating pre-drugs upon irradiation in vivo and thus requires photoreactivity at tissue-penetrating wavelengths. Human tissue absorbs strongly in the u.v. and higher energy visible wavelength range, but is relatively more transparent to longer wavelengths, thus absorption in the red-NIR is ideal for such applications. We have shown that a range of amido ligands may act as the electron donor (and it is likely that other reducible heteroatom ligands may also play this role) and while all the examples we report use a dppz ligand as the electron acceptor it is likely that a wide range of similar conjugated systems could also participate in that context.
Notes and references
- C. Deibel and V. Dyakonov, Rep. Prog. Phys., 2010, 73, 096401 CrossRef.
- M. Riede, T. Mueller, W. Tress, R. Schueppel and K. Leo, Nanotechnology, 2008, 19, 424001 CrossRef CAS PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- M. Gratzel, J. Photochem. Photobiol., C, 2003, 31, 145 CrossRef.
- F. L. Thorp-Greenwood, M. P. Coogan, L. Mishra, N. Kumari, G. Raic and S. Saripellad, New J. Chem., 2012, 36, 64 RSC.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS.
- Y. Jenkins, A. E. Friedman, N. J. Turro and J. K. Barton, Biochemistry, 1992, 31, 10809 CrossRef CAS PubMed.
- E. J. C. Olson, D. Hu, A. Hormann, A. M. Jonkman, M. R. Arkin, E. D. A. Stemp, J. K. Barton and P. F. Barbara, J. Am. Chem. Soc., 1997, 119, 11458 CrossRef CAS.
- J. Fees, W. Kaim, M. Moscherosch, W. Matheis, J. Klima, M. Krejcik and S. Zalis, Inorg. Chem., 1993, 32, 166 CrossRef CAS.
- M. K. Brennaman, T. J. Meyer and J. M. Papanikolas, J. Phys. Chem. A, 2004, 108, 9938 CrossRef CAS.
- K. K. W. Lo and K. H. K. Tsang, Organometallics, 2004, 23, 3062 CrossRef CAS.
- D. Herebian and W. S. Sheldrick, J. Chem. Soc., Dalton Trans., 2002, 966 RSC.
- A. C. Komor, C. J. Schneider, A. G. Weidmann and J. K. Barton, J. Am. Chem. Soc., 2012, 134, 19223 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chem. Acc., 1990, 77, 123 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed , references cited therein.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- J. Jimenez, I. Chakraborty and P. K. Mascharak, Eur. J. Inorg. Chem., 2015, 5021 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1500468. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07125b |
| ‡ For crystallographic data see CCDC 1500468. |
| This journal is © The Royal Society of Chemistry 2016 |