
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability of organic compounds on the oxygen-evolving center of photosystem II and manganese oxide water oxidation catalysts†
Toru
Hayashi
a,
Akira
Yamaguchi
b,
Kazuhito
Hashimoto
c and
Ryuhei
Nakamura
*b
aDepartment of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
bBiofunctional Catalyst Research Team, RIKEN Center for Sustainable Resource Science (CSRS), 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: ryuhei.nakamura@riken.jp
cNational Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
First published on 25th October 2016
Abstract
Surrounding organic residues are indispensable for the high water oxidation activity of the Mn4 cluster in photosystem II. Here, the stability of organic compounds on synthetic Mn oxides was analyzed using inlet electrochemical mass spectroscopy. Carboxyl groups, which are the most abundant residues around the Mn4 cluster, were found to stably facilitate the oxygen evolution reaction.
Efficient and stable non-noble-metal-based catalysts for the oxygen evolution reaction (OER) via water oxidation (2H2O → O2 + 4H+ + 4e−) are highly desirable for the generation of electrons needed to reduce protons or CO2 to produce chemical fuels.1 In nature, the Mn4 cluster (CaMn4O5) of photosystem II (PSII) catalyzes the OER2 with an overpotential of approximately 300 mV1b,3 and a maximum turnover frequency of ∼103 s−1.2d Nine amino-acid residues belonging to the two proteins (D1 and CP43) of PSII are directly coordinated or hydrogen-bonded to the cluster (Fig. 1a) and provide a structural and electrostatic environment that is indispensable for the function and stability of PSII.2a The importance of these amino acids is demonstrated by the fact that any point mutation of the nine residues drastically reduces the activity of the Mn4 cluster for the OER.4 D1-Asp61 has also been regarded as an important residue as a proton acceptor in the concerted proton–electron transfer (CPET) to prevent the charge buildup during the Kok cycle and the starting point of the proton exit pathway,5 which is also shown in Fig. 1a. The residues, however, seem to be at risk for the oxidative decomposition, because the chlorophyll dimer P680 in PSII generates the highest oxidative potential known in biology (1.15–1.26 V vs. SHE)6 for initiating the OER. In fact, it remains uncertain whether the histidine directly liganded to the Mn4 cluster, D1-His332 (Fig. 1a), is subject to one-electron oxidation to a transient radical during the Kok cycle.2c,7 Still, the residues around the Mn4 cluster are highly stable against oxidative decomposition and therefore the turnover number of PSII can reach as high as ∼106,2e even if transient oxidation is operative in this system.
 | ||
| Fig. 1 (a) A schematic diagram of the ligand environment around the Mn4 cluster in PSII adapted from ref. 2a (broken lines: coordination bonds, dotted lines: hydrogen bonds). (b) The synthetic counterpart of PSII examined in this study. The red, blue, and green boxes are shown around carboxyl, imidazolyl, and guanidino groups, respectively. | ||
Recently, several beneficial roles of organic compounds on Mn-based catalysts for the OER at neutral pH were revealed. For example, the coordination of nitrogen-containing organic ligands to the octahedral Mn site of Mn oxides enhances the OER activity,8 a response that is attributed to the increased stabilization of Mn3+, a reaction intermediate of the OER.9 The improved stability of Mn3+ is ascribed to the asymmetric ligand field, where the two eg orbitals of Mn3+ have different energies and therefore the charge disproportionation of Mn3+ to Mn2+ and Mn4+ for eliminating the orbital degeneracy is effectively suppressed.8,10 In addition, pyridine derivatives were shown to enhance the OER activity of Mn oxides by inducing CPET, thereby reducing the required potential for the generation of Mn3+via electrochemical oxidation of Mn2+.11 The enhanced OER activity of Mn oxides modified by amino-acid residues or other ligands was also reported by Najafpour et al.12 The selection of ligands is also of particular importance for the construction of molecular catalysts for the OER.1f However, the synthetic systems described above generally undergo self-oxidation and result in the degradation of organic compounds.
Although organic compounds are indispensable for the efficient OER for both natural and artificial Mn-based catalysts, there is a distinct difference in the stability against the self-oxidation. As determining the origin of the different stability is expected to provide a new design strategy of Mn-based OER catalysts that are highly efficient and stable under neutral conditions, here we analyzed the possible oxidation pathways of organic compounds in a synthetic counterpart and compared them with extant PSII from the viewpoint of energetics.
Russell, Hall, Sauer, and Yachandra proposed that the Mn4 cluster in PSII originated from a Mn mineral.13 Sauer and Yachandra further speculated based on the bond length that the origin was a tunnel-type Mn oxide, such as α-MnO2 with all four Mn cluster arrangements possible in this mineral class (Fig. 1b).13b Fine-tuning of the catalytic activity was also proposed to occur through the reorganization and modification of the Mn oxide mineral during its incorporation into a protein.13b Regardless of the underlying process, the interaction between a complexed Mn compound and the surrounding amino-acid residues would have influenced the evolution of the catalytic ancestor of PSII. In the present study, α-MnO2 and several artificial amino-acid analogs, including benzoate, imidazole, and guanidine, were therefore used as a synthetic model of the reaction center of PSII (Fig. 1b).
Anodic reactions by α-MnO2 electrodes in an electrolyte containing artificial amino-acid analogs were first investigated by measuring the anodic current and the evolution of O2 and CO2 over a range of potentials using a potentiostat, needle-type O2 microsensor, and inlet electrochemical mass spectroscopy (EC-MS) system. Particulate film electrodes were prepared with the synthesized α-MnO2 powder14 using a spray deposition method, as previously described.11 The artificial amino-acid analogs benzoate, imidazole, and guanidine were dissolved in the electrolyte, which consisted of 0.5 M Na2SO4. The pH of the electrolyte was adjusted using NaOH and H2SO4. Detailed experimental conditions are described in the ESI.†
The presence of benzoate in the electrolyte increased both the anodic current and concentration of dissolved O2 generated by the α-MnO2 electrode (Fig. 2a). In the absence of benzoate, the slope of the Tafel plot was 120.7 mV (Fig. 2a-1, inset), indicating that the rate-determining step (RDS) of the OER is a one-electron transfer reaction.15 In addition, the potential at a constant current was not dependent on the pH (Fig. S1, ESI†), indicating that the reaction order of the proton concentration in the total reaction rate was zero. This further suggested that proton transfer was not involved in the RDS or during the pre-equilibrium phase of the step.15 The results from the mechanistic analysis are consistent with previous reports that the RDS of the OER on α-MnO2 is the one-electron oxidation from Mn2+ to Mn3+ without proton transfer.9,11 In contrast, after the addition of benzoate to the electrolyte, the slope value of the Tafel plot decreased (Fig. 2a-1, inset) and the potential at a constant current exhibited pH dependence (Fig. S1, ESI†). The H/D kinetic isotope effect also increased from 1.18 ± 0.09 to 1.45 ± 0.05 (pH(pD) 7; other experimental details are given in the ESI†). These results affirm that the reaction order of the proton concentration increased in the total reaction rate equation by the addition of benzoate.15 The observed enhancement of the OER activity in response to the induction of proton transfer is consistent with our previous finding that CPET is required to avoid the formation of high-energy intermediates (Mn3+–OH2), leading to the enhancement of the activity for the OER.11 Considering the libido rule of general acid–base catalysis,16 which states that the pKa values of bases have to be intermediate between the pKa value of Mn3+–OH2 (0.7) and that of Mn2+–OH2 (10.6) to induce CPET,11,16 benzoate is eligible to be a CPET inducer, as the pKa value is 4.2.17 Further, FT-IR measurement showed the formation of an outer-sphere complex of benzoate and the surface of α-MnO2, a suitable configuration for the induction of CPET (Fig. S2 and S3, ESI†).
 | ||
| Fig. 2 Changes in anodic current density and dissolved O2 concentration for α-MnO2 electrodes by the addition of artificial amino-acid analogs (pH 7.5, scan rate: 1 mV s−1, concentration of benzoate, imidazole (deprotonated form), and guanidine: 50 mM). Inset: Tafel plots (slope: 101.4 mV dec−1 (w/benzoate), 120.7 mV dec−1 (w/o)). | ||
The effect of imidazole on the OER was also examined (Fig. 2b). In the presence of imidazole, a 250 mV negative shift in the onset potential of the anodic current (Eonset), defined as the potential at a current density of 0.5 mA cm−2, was observed for the α-MnO2 electrode (Fig. 2b-1). However, the magnitude of the onset potential shift of the O2 production monitored by the O2 microsensor was only 130 mV (at 0.2 ppm), which was substantially smaller than the shift in Eonset (Fig. 2b-2). The shift in the onset potential of the O2 production was also smaller than that of Eonset when histidine was added to the electrolyte (Fig. S4, ESI†). The large difference in the potential shift for the onset of anodic current and O2 production implied that the oxidation of imidazole and histidine proceeded in a more negative potential region than that for the OER.
To further examine the reason for this difference, in situ EC-MS for the simultaneous monitoring of the anodic current and mass signals of volatile products, and UV-vis measurements were conducted. During an anodic potential scan of α-MnO2 in the presence of imidazole, increases in the ion current for O2 (m/z = 32) were detected with simultaneous production of CO2 (m/z = 44) at the onset potential of approximately 1.25 V (vs. SHE) (Fig. 3a-2 and a-3). No other volatile products, such as NO and NO2, were detected in the EC-MS analysis. However, the onset potential of the anodic current was approximately 0.8 V, which was 250 mV more negative than that of the ion current for O2 (Fig. 2b and 3a). These results demonstrated that the Coulombic efficiency for the OER between 0.8 and 1.25 V was determined to be zero. In the absence of imidazole, the ion current for O2 accompanied by the anodic current and CO2 was not detected (Fig. 3b). Furthermore, a decrease in the time-dependent in situ UV-vis absorption of α-MnO2 electrodes in contact with the electrolyte supplemented with imidazole at resting potential, centered at 510 nm, was observed (Fig. S5, ESI†), with new FT-IR peaks assignable to the oxidation products of imidazole (Fig. S6, ESI†), indicating that electrons injected from imidazole to α-MnO2 were used to reduce Mn4+ to form soluble Mn2+ ions. These results are consistent with the expectation that, although imidazole enhanced OER, the oxidation of imidazole proceeded in a more negative potential region than that for the OER with the concurrent dissolution of Mn2+.
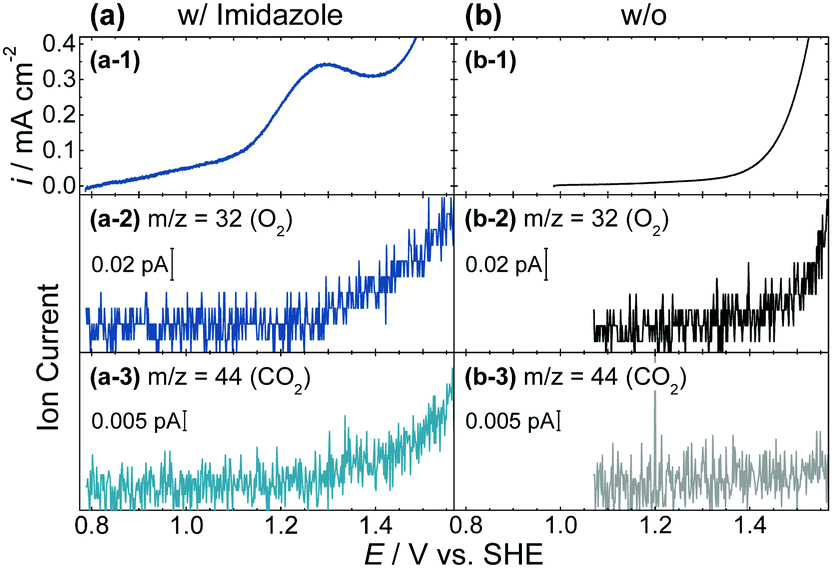 | ||
| Fig. 3 (a-1 and b-1) Current density and (a-2, 3 and b-2, 3) mass signals during the anodic potential scan of α-MnO2 electrodes before and after the addition of 50 mM of the deprotonated form of imidazole (pH 7.5, scan rate: 0.5 mV s−1, unstirred). | ||
In contrast to benzoate and imidazole, the addition of guanidine to the electrolyte resulted in a negligible difference in both the anodic current and OER (Fig. 2c). This finding is consistent with the fact that the pKa value of guanidine is 13.6,17 which exceeds the maximum permissible value to function as a CPET inducer. It also showed that the oxidation of guanidine was negligible in the potential range for the measurement.
The electrochemical analysis of the function of amino-acid analogs using the synthetic counterpart of PSII revealed that benzoate enhanced the OER activity by inducing CPET (Fig. 2a and Fig. S1, ESI†). Although imidazole also enhanced the OER activity, oxidation of imidazole and dissolution of α-MnO2 severely competed with the OER (Fig. 2b and 3), a property that is in sharp contrast with the stable existence of the histidine ligands, D1-His332, and the catalytic core of the Mn4 cluster in PSII (Fig. 1a).
To better understand the reasons for the stability differences of the systems composed of imidazolyl group and α-MnO2 or the Mn4 cluster, we constructed pH-dependent energy diagrams of water and amino acid oxidation for these systems (Fig. 4). The constructed diagrams include the equilibrium potential of the OER, the potential of the α-MnO2 electrodes at 10 μA cm−2 (Eonset′) in the presence of imidazole or histidine measured in the present study, the redox potential of the one-electron oxidation (E1) of histidine in aqueous solution determined in a previous pulse radiolysis study,18 and the reported E1 values of tyrosine in aqueous solution,19 Mn4 cluster (E1(Si+1/Si)),2d,3 redox-active tyrosine residue D1-Tyr161 (YZ),2d,3,20 and P6806 in PSII. To our knowledge, the experimental E1 value of imidazole has not been reported to date. However, this value can be estimated from the reported experimental value of vertical ionization energy (VIE).22 Details of this calculation are provided in the ESI.†
 | ||
| Fig. 4 Thermodynamic potential values related to the oxidation of imidazole and histidine (a) on α-MnO2 and (b) around the Mn4 cluster. The presented values included those experimentally obtained in this study and reported in the literature.2d,3,6,18–20 (Potential vs. SHE was calculated using the reported potential of SHE vs. vacuum, +4.28 V.21) | ||
Based on the pH-dependent energy diagram, it was found that E1 of histidine is approximately 300 mV more positive than Eonset′ in the presence of histidine (Fig. 4a) at pH values ranging from 6 to 8. E1 of imidazole was also estimated to be more positive than Eonset′ in the presence of imidazole (ESI†), suggesting that the one-electron oxidation reaction of histidine and imidazole is thermodynamically unfavorable on α-MnO2 at neutral pH. As Eonset′ in the presence of histidine and imidazole showed pH dependence, it appears that the experimentally observed decomposition of histidine and imidazole on α-MnO2 (Fig. 2b, 3 and Fig. S4, ESI†) is a consequence of a proton-coupled multi-electron oxidation reaction, which results in the simultaneous dissolution of Mn2+ (Fig. S5, ESI†).
Then, how is the oxidation of imidazolyl groups effectively prevented in PSII? In PSII, a potential cascade exists between P680 and the Mn4 cluster, with YZ serving as an intermediate (Fig. 4b). In addition, the E1 values of aromatic amino acids such as tryptophan and tyrosine are predicted to be more positive in protein environments compared to aqueous solutions because of the low dielectric constant of the former (∼3–5 in PSII)2g and the lack of effective protonic contact with the bulk solution which allows deprotonation.20,23 Consistent with this prediction, the E1 value of YZ was found to be approximately 100–200 mV more positive than that of tyrosine in aqueous solution, as shown in Fig. 4a. If the same mechanism holds true for the E1 value of imidazolyl groups in protein environments, the generation of transient histidine radicals via the one-electron oxidation of D1-His332 (Fig. 1a) may also be thermodynamically unfavorable.
The present study also revealed that benzoate functions as an efficient and stable activator of the OER catalyzed by α-MnO2 at neutral pH (Fig. 2a). In PSII, six of the seven direct ligands of the Mn4 cluster are carboxyl groups (Fig. 1a).2a Also, the carboxyl group of D1-Asp61 has been considered to act as a proton acceptor in the CPET in the Kok cycle.5 This exclusive utilization of carboxyl groups is a distinct feature of this enzyme from others. For example, the oxygen reduction reaction (ORR), which is the reverse reaction of the OER, is catalyzed by Cu and Fe coordinated by thiol, imidazolyl groups, or porphyrin nitrogen, but not by carboxyl groups.24 Therefore, it appears that, in the course of evolution, the carboxyl residues were adapted by PSII because of their high durability against self-oxidation during the OER. The introduction of carboxyl groups represents an intriguing strategy to develop stable Mn-based OER catalysts that are highly active under neutral conditions.
In summary, we analyzed the effects and the stability of the organic compounds in a synthetic counterpart of PSII. The comparison of energy diagram of the OER catalyzed by PSII and the synthetic counterpart suggested that imidazolyl groups are oxidized at the surface of α-MnO2 to generate soluble Mn2+ ions via a proton-coupled multi-electron oxidation pathway, while both single and multi-electron oxidation reactions of D1-His332 are effectively inhibited in PSII. In addition, the presence of carboxyl groups, which are abundant around the Mn4 cluster,2a was found to stably facilitate the OER activity of α-MnO2 by inducing the CPET reaction. These findings are expected to provide a new design rationale of organic ligands, which is crucial for the development of functional analogues of the Mn4 cluster for efficient and stable water splitting at neutral pH.
We thank Dr K. Kamiya (Osaka Univ.) for providing access to the EC-MS equipment and K. Jin (Seoul Natl. Univ.) for advice on measuring Tafel plots. This work was supported by JSPS Grant-in-Aid for Scientific Research no. 26288092. T. H. acknowledges Grant-in-Aid for JSPS Fellows no. 15J10583.
Notes and references
- (a) A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed; (b) H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS; (c) Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed; (d) F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920 RSC; (e) K. Jin, A. Chu, J. Park, D. Jeong, S. E. Jerng, U. Sim, H.-Y. Jeong, C. W. Lee, Y.-S. Park, K. D. Yang, G. K. Pradhan, D. Kim, N.-E. Sung, S. H. Kim and K. T. Nam, Sci. Rep., 2015, 5, 10279 CrossRef PubMed; (f) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863 CrossRef PubMed.
- (a) Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 472, 55 CrossRef PubMed; (b) M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Nature, 2015, 517, 99 CrossRef CAS PubMed; (c) J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175 CrossRef CAS PubMed; (d) D. J. Vinyard, M. Gennady, G. M. Ananyev and G. C. Dismukes, Annu. Rev. Biochem., 2013, 82, 577 CrossRef CAS PubMed; (e) N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804 CrossRef CAS PubMed; (f) K. Ogata, T. Yuki, M. Hatakeyama, W. Uchida and S. Nakamura, J. Am. Chem. Soc., 2013, 135, 15670 CrossRef CAS PubMed; (g) P. E. M. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003 CrossRef CAS PubMed.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861 CrossRef CAS PubMed.
- (a) P. J. Nixon and B. A. Diner, Biochemistry, 1992, 31, 942 CrossRef CAS PubMed; (b) R. J. Debus, K. A. Campbell, D. P. Pham, A. M. A. Hays and R. D. Britt, Biochemistry, 2000, 39, 6275 CrossRef CAS PubMed; (c) H. A. Chu, A. P. Nguyen and R. J. Debus, Biochemistry, 1995, 34, 5859 CrossRef CAS PubMed; (d) H. J. Hwang, P. Dilbeck, R. J. Debus and R. L. Burnap, Biochemistry, 2007, 46, 11987 CrossRef CAS PubMed.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284 CrossRef CAS PubMed.
- (a) F. Rappaport, M. Guergova-Kuras, P. J. Nixon, B. A. Diner and J. Lavergne, Biochemistry, 2002, 41, 8518 CrossRef CAS PubMed; (b) M. Grabolle and H. Dau, Biochim. Biophys. Acta, Bioenerg., 2005, 1708, 209 CrossRef CAS PubMed; (c) T. Shibamoto, Y. Kato, M. Sugiura and T. Watanabe, Biochemistry, 2009, 48, 10682 CrossRef CAS PubMed.
- (a) N. Cox and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1020 CrossRef CAS PubMed; (b) A. Boussac, J.-L. Zimmermann, A. W. Rutherford and J. Lavergne, Nature, 1990, 347, 303 CrossRef CAS; (c) B. J. Hallahan, J. H. A. Nugent, J. T. Warden and M. C. W. Evans, Biochemistry, 1992, 31, 4562 CrossRef CAS PubMed; (d) A. Boussac and A. W. Rutherford, Biochemistry, 1992, 31, 7441 CrossRef CAS PubMed; (e) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705 CrossRef CAS PubMed; (f) M. L. Gilchrist, Jr., J. A. Ball, D. W. Randall and R. D. Britt, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 9545 CrossRef; (g) X.-S. Tang, D. W. Randall, D. A. Force, B. A. Diner and R. D. Britt, J. Am. Chem. Soc., 1996, 118, 7638 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153 CrossRef CAS PubMed.
- (a) T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519 CrossRef CAS PubMed; (b) T. Takashima, A. Yamaguchi, K. Hashimoto, H. Irie and R. Nakamura, Electrochemistry, 2014, 82, 325 CrossRef CAS.
- I. I. Mazin, D. I. Khomskii, R. Lengsdorf, J. A. Alonso, W. G. Marshall, R. M. Ibberson, A. Podlesnyak, M. J. Martínez-Lope and M. M. Abd-Elmeguid, Phys. Rev. Lett., 2007, 98, 176406 CrossRef.
- A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Nat. Commun., 2014, 5, 4256 CAS.
- (a) M. M. Najafpour, M. Z. Ghobadi, B. Haghighi, T. Tomo, R. Carpentier, J.-R. Shen and S. I. Allakhverdiev, Plant Physiol. Biochem., 2014, 81, 3 CrossRef CAS PubMed; (b) M. M. Najafpour, M. Z. Ghobadi, D. J. Sedigh and B. Haghighi, RSC Adv., 2014, 4, 39077 RSC; (c) M. M. Najafpour, D. J. Sedigh, S. M. Hosseini and I. Zaharieva, Inorg. Chem., 2016, 55, 8827 CrossRef PubMed.
- (a) M. J. Russell and A. J. Hall, presented in part at Sixth International Congress on Carbon Dioxide Utilization, Breckenridge, Colorado, September, 2001; (b) K. Sauer and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8631 CrossRef CAS PubMed.
- (a) R. M. McKenzie, Mineral. Mag., 1971, 38, 493 CAS; (b) M. Singh, D. N. Thanh, P. Ulbrich, N. Strnadová and F. Štěpánek, J. Solid State Chem., 2010, 183, 2979 CrossRef CAS.
- (a) E. Gileadi, Electrode kinetics for chemists, chemical engineers, and materials scientists, Wiley-VCH, New York, 1993 Search PubMed; (b) J. O'M. Bockris, J. Chem. Phys., 1956, 24, 817 CrossRef.
- W. P. Jencks, J. Am. Chem. Soc., 1972, 94, 4731 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 2014 Search PubMed.
- S. Navaratnam and B. J. Parsons, J. Chem. Soc., Faraday Trans., 1998, 94, 2577 RSC.
- A. Harriman, J. Phys. Chem., 1987, 91, 6102 CrossRef CAS.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004 CrossRef CAS PubMed.
- (a) C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2006, 110, 16066 CrossRef CAS PubMed; (b) A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- B. Jagoda-Cwiklik, P. Slavíček, L. Cwiklik, D. Nolting, B. Winter and P. Jungwirth, J. Phys. Chem. A, 2008, 112, 3499 CrossRef CAS PubMed.
- C. Tommos, J. J. Skalicky, D. L. Pilloud, A. J. Wand and P. L. Dutton, Biochemistry, 1999, 38, 9495 CrossRef CAS PubMed.
- (a) S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936 CrossRef CAS PubMed; (b) S. A. Roberts, A. Weichsel, G. Grass, K. Thakali, J. T. Hazzard, G. Tollin, C. Rensing and W. R. Montfort, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental conditions, supplementary figures and discussion. See DOI: 10.1039/c6cc07092b |
| This journal is © The Royal Society of Chemistry 2016 |