
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of terminal 3′-hydroxymethyl modification of an RNA primer on nonenzymatic primer extension†
Ayan
Pal‡
ab,
Rajat S.
Das‡
 cd,
Weicheng
Zhang‡
ae,
Megan
Lang
c,
Larry W.
McLaughlin
c and
Jack W.
Szostak
*abe
cd,
Weicheng
Zhang‡
ae,
Megan
Lang
c,
Larry W.
McLaughlin
c and
Jack W.
Szostak
*abe
aHoward Hughes Medical Institute, Department of Molecular Biology and Center for Computational and Integrative Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, Massachusetts 02114, USA. E-mail: szostak@molbio.mgh.harvard.edu
bDepartment of Genetics, Harvard Medical School, 77 Avenue Louis Pasteur, Boston, Massachusetts 02115, USA
cBoston College, Department of Chemistry, Merkert Chemistry Center, 2609 Beacon Street, Chestnut Hill, Massachusetts 02467, USA
dAlnylam Pharmaceuticals, 300 Third Street, Cambridge, Massachusetts 02142, USA
eDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford St., Cambridge, Massachusetts 02138, USA
First published on 5th September 2016
Abstract
The significance of the precise position of the hydroxyl at the 3′-end of an RNA primer for nonenzymatic template-directed primer extension is not well understood. We show that an RNA primer terminating in 3′-hydroxymethyl-2′,3′-dideoxy-guanosine has greatly diminished activity, suggesting that the spatial preorganization of the terminal sugar contributes significantly to the efficiency of primer extension.
Template-directed primer extension with chemically activated ribonucleotide monomers, such as nucleotide 5′-phosphorimidazolides, has been extensively studied as a model for nonenzymatic RNA replication during the origin of life.1–4 In this model, activated nucleotide monomers first interact with the RNA primer–template complex by binding reversibly to complementary sites on the template. Extension of the primer occurs by nucleophilic attack of either the 2′- or 3′-hydroxyl of the 3′-terminal nucleotide of the primer on the activated 5′-phosphate of an adjacent template-bound monomer. A 2′–5′ or 3′–5′ phosphodiester bond is formed when the nucleophile displaces the leaving group on the phosphate of the activated monomer. This reaction is accelerated by the catalytic assistance of a divalent cation, such as Mg2+ or Mn2+, although it is not known whether the metal ion interacts with the nucleophilic hydroxyl, the phosphate, or both. Various leaving groups on the phosphate of the incoming monomers, such as imidazole,5 2-methylimidazole,6 and oxyazabenzotriazole,7 have been studied in the primer extension reaction. Oligomerization on mineral surfaces has also been carried out using 1-methyladenine8 as the leaving group. Markedly improved polymerization, in terms of both rate and 3′-5′ regioselectivity, is achieved with 2-methylimidazole9 as the leaving group at a pH of 8 to 8.5, although hydroxyazabenzotriazole gives faster rates at pH 8.9–9.5.10
Previous studies have demonstrated that primer extension is more efficient on A-form templates such as RNA compared to B-form templates such as DNA.11,12 Moreover, our laboratory has shown that the sugar pucker of activated ribonucleotides switches from mostly 2′-endo in the free state to mostly 3′-endo upon binding to an RNA template.13 These observations suggest that an A-form primer–template complexed with activated ribonucleotides monomers in the 3′-endo conformation represents the optimal conformation for in-line attack of the nucleophile at the 3′ end of the primer on the 5′-phosphate of the adjacent activated monomer. A better nucleophile, such as the 3′-amine of N3′–P5′-linked phosphoramidate DNA (3′-NP-DNA), is known to improve the kinetics of the extension reaction.14,15 However, the importance of the structural position and rigidity of the nucleophile at the 3′-position has not been systematically investigated. Based on the fact that both the 2′ and 3′ hydroxyls are able to attack the phosphate of the incoming nucleotide (to an extent controlled by leaving group and metal ion identity), it might seem that the precise position of the nucleophile is not critical. Furthermore, primers ending in a 2′-deoxynucleotide can still be extended at about 10% of the rate of primer ending in a ribonucleotides,16 despite the higher pKa of the hydroxyl (∼14 vs. 12.5).16,17 The intrinsic sugar preference for the 2′-endo conformation could be countered by the cooperative induction of A-form helical geometry by the remainder of the RNA primer.18 On the other hand, altritol and hexitol monomers, which have different sugar moieties, are poor substrates for continued primer extension, although it is unclear whether this reflects poor positioning of the attacking nucleophile or the electrophilic phosphate.19
To explore the structural requirements for the placement of the nucleophile, we have replaced the terminal ribonucleotide G of a primer by 3′-hydroxymethyl-2′,3′-dideoxy-guanosine, represented here as dG*. Different arguments suggest that this modified 3′-terminus could either enhance or interfere with primer extension. The hydroxymethyl group of dG*, a primary alcohol, should be a better nucleophile than the 3′-hydroxyl of a 2′-deoxynucleotide, because of its lower pKa and decreased steric hindrance (although hydrogen bonding of the hydroxyl to a phosphate oxygen could increase its pKa as well as causing steric problems). On the other hand, the displacement of the hydroxyl of dG* away from the sugar, and its greater conformational flexibility, could decrease the rate of primer extension if precise spatial positioning is a critical factor in the reaction.
In order to investigate the effects of structural perturbation at the 3′-position on the primer extension reaction, we first had to prepare an RNA primer terminating in dG*. Here we describe the synthesis of 5′-(β-cyanoethyl) phosphoramidite of 3′-hydroxymethyl-2′,3′-dideoxy-guanosine and its use in the reverse solid-phase preparation of a primer terminating with dG* (Fig. 1). Several previous reports have described the synthesis of nucleotide analogs with an extended sugar phosphate backbone, typically beginning with a modified abasic sugar or carbocycle20–25 followed by glycosylation to incorporate the purine or pyrimidine nucleobase. This approach leads to the formation of unwanted regioisomers that are difficult to separate.26 In addition, maintenance of the correct stereochemistry at the 3′-carbon has been difficult.27,28 In our study, the 3′-hydroxymethyl-dG phosphoramidite was synthesized, purified, and characterized by a modification of the procedure originally reported by Mesmaeker et al.29 that avoids these problems by 3′-homologation of 2′-deoxyguanosine (Scheme 1).
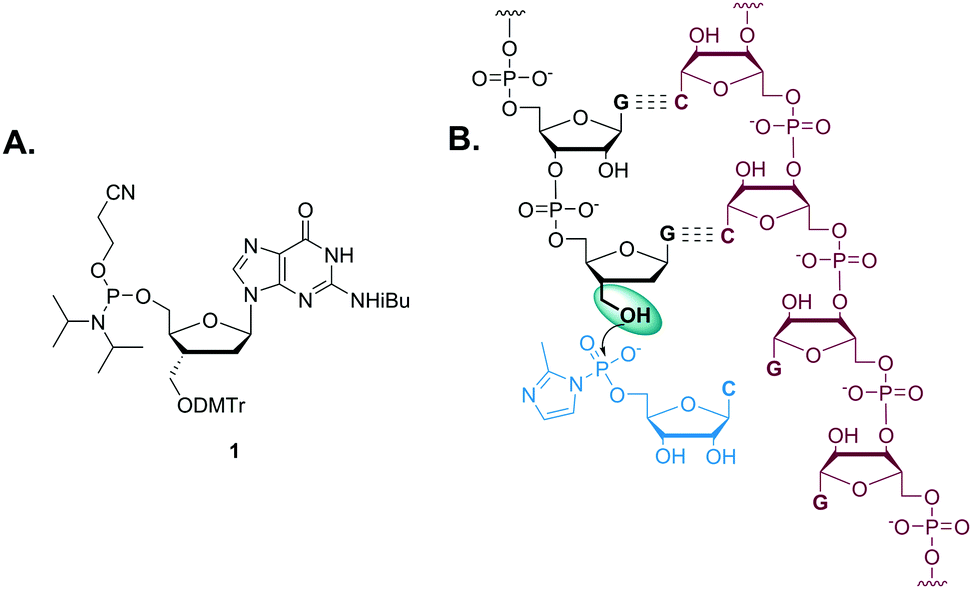 | ||
| Fig. 1 (A) Structure of 5′-(β-cyanoethyl) phosphoramidite of 3′-hydroxymethyl-2′,3′-dideoxy-guanosine. (B) Schematic representation of template-directed extension of 3′-hydroxymethyl-2′,3′-dideoxy-guanosine-terminated primer in the presence of 2-MeImpC. | ||
 | ||
| Scheme 1 Synthesis of phosphoramidite. Reaction conditions: (a) TBDPSCl, DMAP, pyr, 20 °C, 36 h, 92%; (b) o-(p-tolyl)-chlorothionoformate, DMAP, ACN, 20 °C, 1 h, 94%, (c) β-tributylstannylstyrene, AIBN, toluene, 20 to 80 °C, 48 h; (d) OsO4, NMO, dioxane, 20 °C, 2 h; (e) NaIO4, 20 °C, 3 h, 28% over 3 steps; (f) NaBH4, MeOH, 0 to 20 °C, 30 min, 94%; (g) DMTrCl, DMAP, pyr, 20 °C, 12 h, 93%; (h) TBAF, THF, 20 °C, 8 h; (i) 2-cyanoethyl N,N,N,N-tetraisopropylphosphoramidite, tetrazole, DIEA, DCM, 20 °C, 2 h, 89% over 2 steps. | ||
To ensure the stereoselective introduction of the 3′-α-hydroxymethyl functionality, the β-face of the sugar ring was blocked by both the bulky tert-butyl diphenylsilyl group and the guanine nucleobase, so that the attack of an incoming nucleophile would occur predominantly from the α-face. The 3′-hydroxyl was converted into its corresponding o-(p-tolyl)thiocarbonate ester 4. The ester 4 was reacted with β-tributylstannylstyrene in the presence of AIBN to form a new C–C bond via β-elimination of the tributylstannyl group. Since the β-face of the sugar ring is sterically blocked, styryl addition occurs preferentially through the α-face. The crude reaction mixture was filtered through a short silica pad to remove excess reagents and dried. The resulting crude product was used directly in the next step. The styryl group was cleaved by dihydroxylation using osmium tetroxide, followed by oxidation of the diol with sodium periodate to generate a 3′-α-C-formyl group in 5. The aldehyde of 5 was efficiently reduced to yield the hydroxymethyl group of 6 by sodium borohydride. This 3′-α-C-hydroxymethyl group was then protected with DMTr, and the 5′-alcohol of 7 retrieved by desilylation, followed by conversion to phosphoramidite 1 in excellent yield. This reverse amidite 1 was incorporated as the terminal residue of an RNA oligomer by reverse solid-phase synthesis. The oligomer was purified by HPLC and characterized by LC-MS. The dG*-terminated oligomer obtained was at greater than 97% purity with a trace of N-1 oligomer.
To examine the effect of dG* on template-directed nonenzymatic primer extension, we used a 5′-fluorophore tagged dG*-terminated RNA primer (P1, 5′-Cy3-GACUGACUGdG*-3′) complexed with a complementary RNA template strand (T, 3′-CUGACUGACCGGGGAA-5′) containing a homopolymeric G4 site to be copied by the activated monomer, cytidine-5′-phosphor-(2-methyl) imidazolide (2-MeImpC). As a control, a 5′-fluorophore tagged dG-terminated RNA primer (P2, 5′-Cy3-GACUGACUGdG-3′) was also prepared and time courses of primer extension with the two primers were compared.
To determine the optimal conditions for the primer extension reaction, a series of experiments were performed with 1 μM primer, 5 μM template, and 100 mM of different divalent cations including Mg2+, Mn2+ and Ca2+, in the presence or absence of monovalent cation (Na+ or K+) and in the presence of 50 mM of activated monomers and 200 mM buffer (pH from 5.5 to 8.5). Surprisingly, we failed to observe any significant primer extension from the dG* primer P1 even after 1 day under most conditions. The best condition, with 100 mM Mg2+ at pH 7.5, yielded less than 5% extended products after 24 hours, with +3 extended product being the predominant contributor (Fig. 2A). The kobs was approximately 0.012 h−1. We were unable to detect any +1 or +2 extended products, most likely because after the very slow addition of the first monomer, addition of the following two residues is fast as the terminal residue of the +1 or +2 extended products is then a standard ribonucleotide. On the other hand, P2 extended to varying extents depending on pH, metal ions and time (Fig. 2B). At pH 7.5 with Mg2+, full-length +3 products appeared after 2 hours with kobs of ∼2.8 h−1. After 24 hours, more than 95% of the P2 was extended to +3 or +4, in striking contrast to the minimal extent of primer extension of P1.
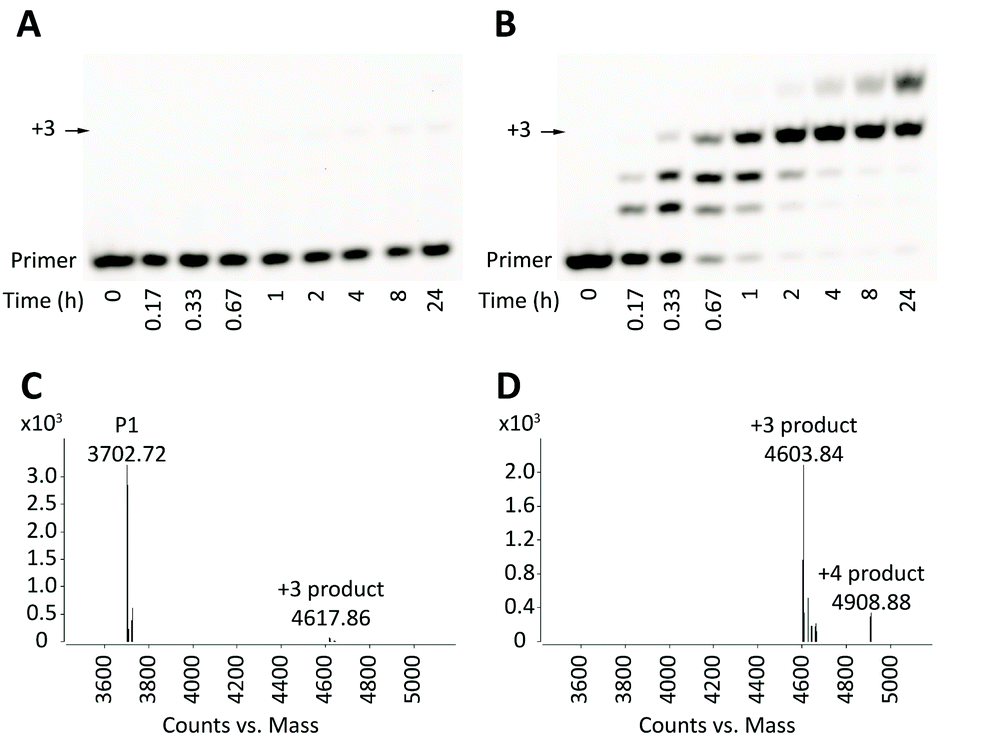 | ||
| Fig. 2 Gel electrophoresis and LC-MS analysis of primer extension reactions. Reaction conditions: 1 μM primer, 5 μM template T, 200 mM HEPES pH 7.5, 100 mM MgCl2, 50 mM 2-MeImpC. (A) and (B) Gel analysis of primer extension of primers P1 and P2 respectively; (C) and (D) LC-MS analysis of primer extension of primers P1 and P2 after 24 hours respectively. | ||
To confirm that the small amount of P1 +3 product observed by PAGE analysis did indeed result from extension from the dG* residue (and not from trace levels of N-1 oligomer), we used LC-MS to analyse the 24 hour time point of the primer extension reaction. While unreacted P1 was the predominant species (Fig. 2C), approximately 3% of primer extended to +3 product (Fig. 2C; see also ESI,† Fig. S1), consistent with the results of the gel electrophoresis analysis. The mass of the P1 +3 product was 14 daltons greater than that of the P2 +3 product, as expected from replacement of the primer 3′-hydroxyl with a 3′-hydroxymethyl group.
In conclusion, template-directed nonenzymatic extension of a primer with a 3′-terminal dG* proceeds very slowly, at a rate less than 1% that of a primer with a terminal dG residue. We suggest that two major factors may cause the slow primer extension from dG*. First, the location of the hydroxyl in dG* is ∼1.5 Å from the position of the normal 3′-hydroxyl. This altered location may make nucleophilic attack of the hydroxyl on the phosphate of the incoming activated nucleotide difficult either due to the altered position per se or due to possible hydrogen bonding of the hydroxyl to a phosphate oxygen, or may hinder coordination with the catalytic Mg2+ ion. Second, the conformational flexibility of the primary hydroxyl group may decrease the probability of the required spatial positioning of the nucleophile for an in-line attack on the 5′-phosphate of the activated monomer. Our results suggest that the normal location of the nucleophile, on the 3′-carbon of the ribose or deoxyribose sugar, is required for efficient nonenzymatic template directed primer extension.
We thank Drs Victor Lelyveld, Wen Zhang, Noam Prywes, Neha Kamat and Mr Travis Walton for helpful discussions. J. W. S. is an Investigator of the Howard Hughes Medical Institute. This work was supported in part by a grant (290363) from the Simons Foundation to J. W. S.
Notes and references
- L. E. Orgel, Crit. Rev. Biochem. Mol. Biol., 2004, 39, 99 CrossRef CAS PubMed.
- T. Wu and L. E. Orgel, J. Am. Chem. Soc., 1992, 114, 5496 CrossRef CAS PubMed.
- T. Wu and L. E. Orgel, J. Am. Chem. Soc., 1992, 114, 7963 CrossRef CAS PubMed.
- S. S. Mansy, J. P. Schrum, M. Krishnamurthy, S. Tobé, D. A. Treco and J. W. Szostak, Nature, 2008, 454, 122 CrossRef CAS PubMed.
- R. Lohrmann and L. E. Orgel, Tetrahedron, 1978, 34, 853 CrossRef CAS.
- G. F. Joyce, T. Inoue and L. E. Orgel, J. Mol. Biol., 1984, 176, 279 CrossRef CAS PubMed.
- P. Hagenbuch, E. Kervio, A. Hochgesand, U. Plutowski and C. Richert, Angew. Chem., Int. Ed. Engl., 2005, 44, 6588 CrossRef CAS PubMed.
- K. J. Prabahar and J. P. Ferris, J. Am. Chem. Soc., 1997, 119, 4330 CrossRef CAS PubMed.
- T. Inoue and L. E. Orgel, J. Mol. Biol., 1982, 162, 201 CrossRef CAS PubMed.
- S. R. Vogel, C. Deck and C. Richert, Chem. Commun., 2005, 4922 RSC.
- M. Kurz, K. Göbel, C. Hartel and M. W. Göbel, Angew. Chem., Int. Ed. Engl., 1997, 36, 842 CrossRef CAS.
- M. Kurz, K. Göbel, C. Hartel and M. W. Göbel, Helv. Chim. Acta, 1998, 81, 1156 CrossRef CAS.
- N. Zhang, S. Zhang and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 3691 CrossRef CAS PubMed.
- S. Zhang, N. Zhang, J. C. Blain and J. W. Szostak, J. Am. Chem. Soc., 2013, 135, 924 CrossRef CAS PubMed.
- S. Zhang, J. C. Blain, D. Zielinska, S. M. Gryaznov and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17732 CrossRef CAS PubMed.
- T. Wu and L. E. Orgel, J. Am. Chem. Soc., 1992, 114, 317 CrossRef CAS PubMed.
- G. I. Birnbaum, J. Giziewicz, C. P. Huber and D. Shugar, J. Am. Chem. Soc., 1976, 98, 4640 CrossRef CAS PubMed.
- M. C. Wahl and M. Sundaralingam, Nucleic Acids Res., 2000, 28, 4356 CrossRef CAS PubMed.
- I. A. Kozlov, M. Zielinski, B. Allart, L. Kerremans, A. Van Aerschot, R. Busson, P. Herdewijn and L. E. Orgel, Chem. – Eur. J., 2000, 6, 151 CrossRef CAS.
- H. Boumchita, M. Legraverend and E. Bisagni, Heterocycles, 1991, 32, 1785 CrossRef CAS.
- J. Wachtmeister, B. Classon, I. Kvarnström and B. Samuelsson, Nucleosides Nucleotides, 1997, 16, 809 CAS.
- J. Wachtmeister, A. Mühlman, B. Classon and B. Samuelsson, Tetrahedron, 1999, 55, 10761 CrossRef CAS.
- H. R. Moon, H. O. Kim, M. W. Chun, L. S. Jeong and V. E. Marquez, J. Org. Chem., 1999, 64, 4733 CrossRef CAS PubMed.
- J. Wachtmeister, C. Björn, S. Bertil and I. Kvarnström, Tetrahedron, 1997, 53, 1861 CrossRef CAS.
- M. Janson, L. Svansson, S. C. T. Svensson, I. Kvarnström, B. Classon and B. Samuelsson, Nucleosides Nucleotides, 1992, 11, 1739 CAS.
- G. S. Buenger and V. E. Marquez, Tetrahedron Lett., 1992, 33, 3707 CrossRef CAS.
- T. Kofoed, P. B. Rasmussen, P. Valentin-Hansen and E. B. Pedersen, Acta Chem. Scand., 1997, 51, 318 CrossRef CAS.
- J. J. Vasseur, F. Debart, Y. S. Sanghvi and P. D. Cook, J. Am. Chem. Soc., 1992, 114, 4006 CrossRef CAS.
- Y. S. Sanghvi, R. Bharadwaj, F. Debart and A. De Mesmaeker, Synthesis, 1994, 1163 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc06925h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |