
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of the carbon–chalcogen (S, Se, Te) bond at the 2,6-positions of BODIPY via Stille cross-coupling reaction†
E.
Palao
,
T.
Slanina
and
P.
Klán
*
Department of Chemistry and RECETOX, Faculty of Science, Masaryk University, Kamenice 5, 625 00, Brno, Czech Republic. E-mail: klan@sci.muni.cz
First published on 16th September 2016
Abstract
Seven new 2-chalcogen- or 2,6-dichalcogen- (S, Se, Te) BODIPY derivatives were synthesized in good to excellent yields (55–95%) by a Pd-catalyzed C–heteroatom Stille cross-coupling reaction, overcoming the limitations of SNAr. The fluorophores show interesting tunable optical properties associated with the formation of a twisted intramolecular charge transfer excited state and competing intersystem crossing.
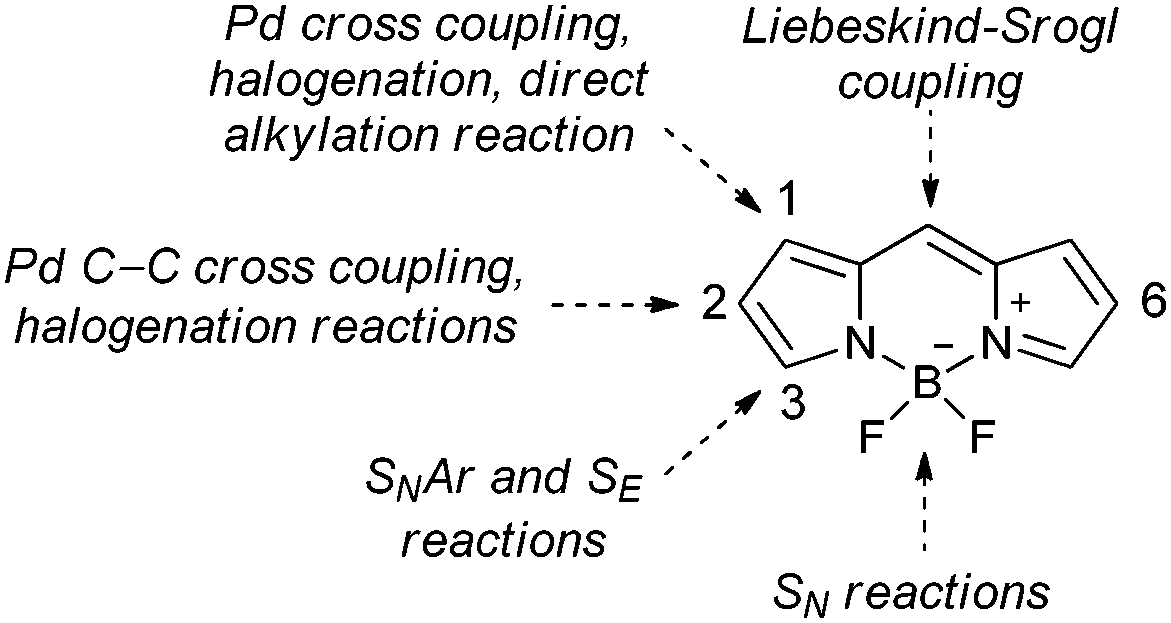
Due to their remarkable photophysical properties and simple synthesis, boron-dipyrromethene dyes (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene, BODIPY)1 have found use in many applications such as laser dyes,2 molecular probes,3 triplet photosensitizers,4 or photoremovable protecting groups.5–7 BODIPY derivatives can be synthesized by simple condensation of suitable reaction partners or by modification of the BODIPY scaffold (Fig. 1).8 The most successful BODIPY synthetic functionalization includes SEAr reactions, such as halogenations at the 2/6-positions,9–12 nucleophilic aromatic substitutions at the 1/3/5/7/8-positions,8 nucleophilic substitutions at boron,13 or metal-catalyzed C–C cross coupling at all positions.14
 | ||
| Fig. 1 Typical synthetic routes for BODIPY derivatization. | ||
Despite numerous synthetic methods available for the BODIPY functionalization, there are still some limitations. The lowest coefficients in the LUMO at the 2,6-positions of BODIPY15 hamper nucleophilic substitution which is normally used for installation of heteroatom-containing substituents. Therefore, BODIPY derivatives substituted with chalcogen atoms at the 2,6-positions, such as ethers,16 amines,17 or thioethers,18,19 are rare. We recently reported on the synthesis of several 3-chalcogen substituted BODIPYs, which exhibit intersystem crossing (ISC) quantum yields comparable to those of iodo-analogues.20 Due to their high photostability and low toxicity, chalcogen-substituted BODIPYs are promising triplet sensitizers21 which can be used in photodynamic therapy22 or triplet–triplet annihilation upconversion.4,23
Ortiz and coworkers have recently demonstrated that heavy-atom substituents at the 2,6-positions have the highest impact on ISC quantum yields.11 When we decided to prepare 2,6-chalcogeno-BODIPY derivatives, we found that a standard nucleophilic substitution reaction to install a chalcogen-containing substituent at these positions cannot be used. Although Pd-catalyzed C–C bond formation via cross coupling at the these positions is common, metal-catalyzed cross-coupling reactions to form the C–N, C–O or C–S bonds, such as Buchwald–Hartwig and Ullmann reactions, have not yet been reported. Recently, Knight and coworkers reported on a Cu-catalyzed amination of 2-iodo BODIPYs.24 Interestingly, instead of 2-amino derivatives, 3-amino BODIPYs were formed via a formal cine-substitution. Highly nucleophilic amines were not involved in a direct nucleophilic substitution of iodine but preferentially attacked the 3-position. Metal-catalyzed reactions on 2,6-halogen substituted BODIPYs, such as Negishi25 and Hiyama cross couplings, have recently been hypothesized.12
For this work, we chose a different strategy for the synthesis of 2- or 2,6-chalcogeno-BODIPYs using a Stille-type Pd-catalyzed reaction with organostannanes, which has already been used to form chalcogen–carbon bonds in some other aromatic and heteroaromatic systems.26–29 In addition, steady-state and transient absorption spectroscopies were used to characterize the excited states of the prepared derivatives.
The first monosubstituted 2-phenylselanyl BODIPY derivative 3a was synthesized from 6-iodo derivative 120 using phenyl tributylstannyl selenide 2a30 and Pd(PPh3)4 in toluene under an argon atmosphere in 75% yield (Scheme 1). Our original objective was to explore the reactivity of a BODIPY derivative which has no substituents in the vicinity of the C–Se bond (the 1/3-positions) to make sure that cine-substitution24 does not occur. No traces of any cine-substitution product were found; compound 3a was formed as the only detectable product. Substituted trialkylstannanes were used as suitable sources of the chalcogen-containing group because they readily undergo ligand exchange in Pd-catalyzed reactions.26 To the best of our knowledge, this reaction represents the first example of a C–heteroatom cross coupling reaction on a BODIPY core. We further expanded the scope of this method, and compounds 3b and 3c were synthesized from phenyl (2b31) and benzyl (2c32) tributylstannyl sulfides, respectively, in high yields (Scheme 1).
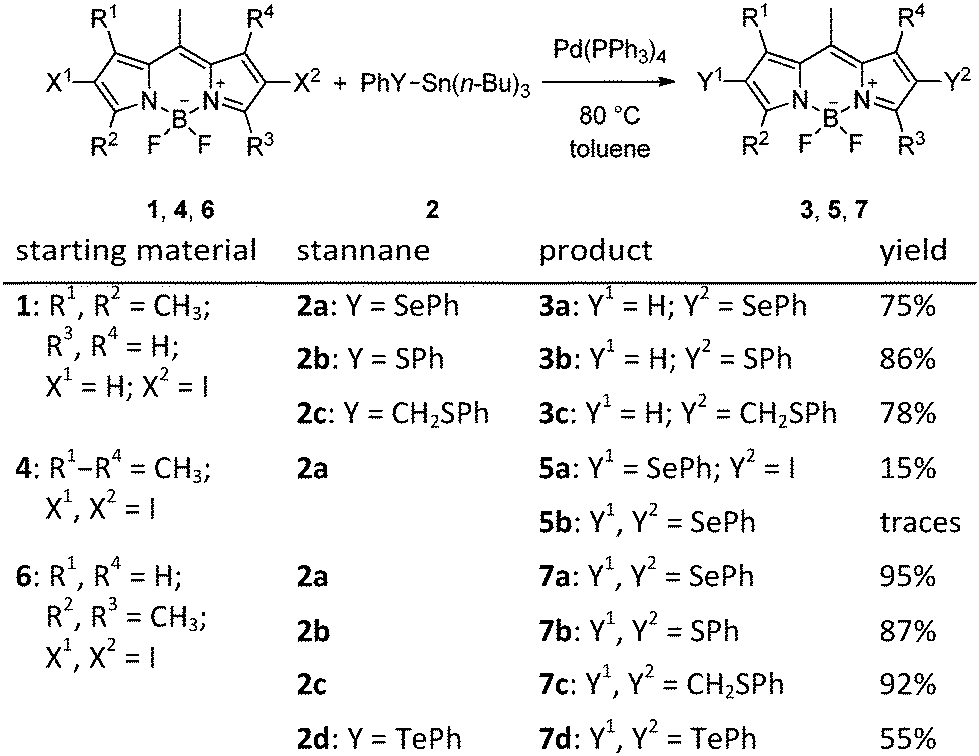 | ||
| Scheme 1 Synthesis of the BODIPY derivatives 3, 5, 6 and 7. | ||
In the next step, we attempted to prepare 2,6-dichalcogen-substituted BODIPYs using the same method. Compound 4 was chosen as a precursor because it has already been successfully used for C–C cross-couplings reactions9 and can be synthesized from a commercially available BODIPY derivative. However, the mono-substituted derivative 5a was isolated in poor yields (8–15%) as the major product, and the disubstituted BODIPY 5b was detected only in traces by NMR in a mixture with 5a even after a careful reaction optimization (the starting material was always recovered; Table S1, ESI†). We hypothesized that steric factors (the methyl groups in the 1,7-positions) may hinder the coupling reaction in the 2,6-positions. Thus, a less substituted BODIPY derivative 633 was used in the synthesis of three new disubstituted derivatives 7a–c in high yields (87–95%) under the same reaction conditions used in the synthesis of derivatives 3 (Table 1). In addition, an analogous reaction of the organostannane 2d34 gave the ditelluro-substituted BODIPY 7d in a moderate yield (55%) despite several optimization efforts.
| Compound | λ absmax/nm | ε max |
λ
fmax
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) /nm
/nm |
Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) c/cm−1 c/cm−1 |
Φ f /% | τ f /ns | Φ ISC | Φ Δ | Φ decomp /10−5 |
|---|---|---|---|---|---|---|---|---|---|
| a ε max/mol−1 dm3 cm−1. b 1: λexc = 460 nm; 3a–c: λexc = 460 nm 6: λexc = 510 nm; 7a–d: λexc = 490 nm. c Stokes shift. d Fluorescence (Φf) and singlet oxygen generation (ΦΔ) quantum yields; the standard deviation (SD) was calculated from 5 measurements. e Fluorescence lifetimes (τf) and photodecomposition quantum yields (Φdecomp); the SD was calculated from 3 measurements. f ISC quantum yields; the SD was calculated from 6 measurements. | |||||||||
| 1 | 493 | 35400 |
520 | 1053 | 4.8 ± 0.2 | 0.53 ± 0.01 | 0.56 ± 0.03 | 0.53 ± 0.02 | 24 ± 2 |
| 3a | 483 | 45292 |
642 | 5128 | 1.9 ± 0.1 | 5.75 ± 0.01 | 0.57 ± 0.02 | 0.55 ± 0.01 | 4.6 ± 0.5 |
| 3b | 482 | 36950 |
633 | 4949 | 13.7 ± 0.3 | 2.96 ± 0.05 | 0.15 ± 0.01 | 0.15 ± 0.02 | 3.5 ± 0.5 |
| 3c | 489 | 33629 |
623 | 4400 | 10.1 ± 0.3 | 6.10 ± 0.1 | 0.15 ± 0.01 | 0.14 ± 0.01 | 20 ± 1 |
| 6 | 535 | 78378 |
553 | 608 | 4.5 ± 0.1 | 0.47 ± 0.05 | 0.49 ± 0.01 | 0.52 ± 0.03 | 16 ± 1 |
| 7a | 518 | 48934 |
642 | 3729 | 2.2 ± 0.1 | 3.13 ± 0.02 | 0.46 ± 0.01 | 0.42 ± 0.03 | 13 ± 1 |
| 7b | 519 | 54528 |
642 | 3692 | 14.9 ± 0.2 | 3.22 ± 0.08 | 0.07 ± 0.01 | 0.07 ± 0.01 | 1.7 ± 0.2 |
| 7c | 524 | 50723 |
640 | 3459 | 29.0 ± 0.5 | 4.90 ± 0.07 | 0.10 ± 0.02 | 0.09 ± 0.01 | 12 ± 1 |
| 7d | 519 | 78048 |
642 | 3692 | 18.3 ± 0.2 | 2.91 ± 0.03 | 0.07 ± 0.01 | 0.07 ± 0.01 | 1.0 ± 0.1 |
The absorption spectra of the BODIPY derivatives 1, 3a–c, 6 and 7a–d in acetonitrile possessed a major narrow absorption band with the absorption maximum (λabsmax) in the region of 482–535 nm (Table 1; Fig. S25, ESI†). The λabsmax values for the mono-substituted derivatives 1 and 3a–c were shifted hypsochromically (λabsmax = 482–493 nm) compared to those of the disubstituted derivatives 6 and 7a–d (516–535 nm) and were only weakly dependent on the solvent polarity (Fig. S26, ESI†). The absorption band broadening observed for aqueous solutions (water/acetonitrile, 99:1; Fig. S26, ESI†) indicates aggregation35 of the dye molecules. All prepared compounds exhibited relatively weak fluorescence (Φf = 1.9–29%); the S- and Te-BODIPY analogues were the brightest fluorophores in the series. Furthermore, phosphorescence from 7a in frozen hexane and acetonitrile was detected (λpmax = 749 nm; Fig. S40, ESI†); the phosphorescence spectrum corresponds to those of halogen-substituted BODIPYs reported before.36
Non-substituted BODIPYs usually exhibit fluorescence with a lifetime of ∼5 ns and negligible ISC quantum yields, and their emission originates from the locally excited (LE) state.20,37,38 Lower Φf values were anticipated especially for the Se-, Te- and I-derivatives as a result of spin–orbit coupling between singlet and triplet states (the heavy-atom effect, HAE).11,20,21 Indeed, the fluorescence lifetime of iodo-BODIPYs 1 and 6 was shortened by one order of magnitude (τf ≈ 0.5 ns). These compounds exhibited sharp emission bands and small Stokes shifts (Δ = 600–1000 cm−1; Fig. S27, ESI†), and we identified the LE and T1 (observed only at 77 K; Fig. S46, ESI†) states as the only emissive states.
On the other hand, the chalcogen-substituted BODIPYs displayed broad emission bands and large Stokes shifts (Δ = 3692–5128 cm−1; Fig. S28–S35, ESI†), fluorescence lifetimes (τf ≈ 3–6 ns) in the same order as those of unsubstituted BODIPYs,20 and surprisingly high Φf values, especially in the case of ditelluro-analogue 7d (18%). BODIPY derivatives possessing large Stokes shifts based on geometrical changes upon excitation,39 intramolecular charge transfer (ICT),40 or extension of the chromophore41 have already been reported. Thus the spectroscopic properties of derivatives 3 and 7 suggested that the nature of their emissive electronic state differs from those of iodo-derivatives 1 and 6.
Such a spectroscopic behavior has already been observed for BODIPYs consisting of electron donor and acceptor units, and it was suggested that an emissive twisted intramolecular charge transfer excited state (TICT) is formed.35,42 This state is formed by redistribution of the electronic density due to excited-state geometry relaxation, such as rotation of a bond connecting the donor and acceptor units, and can be indicated by high Stokes shifts.42–44 Despite the fact that TICT states have a high susceptibility to various non-radiative quenching processes,35 several examples of TICT fluorescence are available in the literature.45,46 Due to its higher dipole moment, a TICT state is stabilized in more polar solvents35,46 and at lower temperatures46 (exhibited by bathochromic shifts of the emission maxima). On the other hand, the LE state is known to be pronounced in less polar solvents and frozen solutions where any substantial reorganization of neither the solvent nor chromophore geometry can take place.47
We chose 7a to explore the origin of the observed broad, red-shifted emission bands in the chalcogeno-BODIPY derivatives. The emission band with λfmax = 622 nm in hexane shifted to 642 and 666 nm in acetonitrile and water, respectively (Fig. S36, ESI†). This effect is smaller than that of fluorophores allowing for a higher charge separation35,48 but still significant for a compound with an electron donor (Se) directly attached to the chromophore.47 Upon cooling the solutions of 7a in both acetonitrile (293 → 223 K) and hexane (293 → 180 K), bathochromic shifts of its emission maxima by approximately 8 nm were observed (Fig. S37 and S38, ESI†). Upon freezing a hexane solution at 77 K, dual fluorescence appeared: the intensity of the band at 620 nm decreased and a new emission signal at ∼560 nm appeared (Fig. S39, ESI†). We assign this new sharp band to the π,π* LE state, whereas a still well-detected, bathochromically shifted broad band is assigned to the TICT state.
To evaluate the effect of chalcogen substituents as heavy atoms, the quantum yields of ISC (ΦISC) were obtained by ns transient absorption spectroscopy20,49 for all prepared derivatives and compared with the quantum yields of singlet oxygen generation (ΦΔ; Table 1), a common tool for determination of ISC efficiency in BODIPY derivatives.50 All triplet–triplet absorption spectra (Fig. S41–S43, ESI†) are in accord with those obtained for other reported BODIPYs.20,50 Practically, the same ΦISC and ΦΔ values found here indicate that there are no competing pathways of triplet deactivation. Table 1 shows that ISC is the most efficient in the iodo- (1 and 6) and phenylseleno- (3a and 7a) derivatives. As sulfur is the ‘least heavy’ member of chalcogens, a rather inefficient ISC in 7b and 3b was expected. On the other hand, a small ΦISC in the Te-derivative 7d is at variance with our recent study on 3-substituted BODIPYs, in which ΦISC increased in the order of the H < Cl < Se/I < Te substitution in the 3-position.20 The ΦISC values were only slightly lower for the disubstituted derivatives than those of the mono-substituted analogues.
The energy of a TICT state can be obtained directly from the emission spectra or estimated from redox potentials of the donor (chalcogen) and acceptor (BODIPY) moieties, treated as a pair of radicals at a fixed distance.51 The ground-state reduction potentials of PhYMe˙+/PhYMe in acetonitrile are +1.56, +1.22, and +0.77 V vs. Ag/AgCl for Y = S, Se, and Te, respectively,52 whereas that of the excited (LE) BODIPY core estimated from the ground state reduction potential (E[BODIPY/BODIPY˙−] = −0.73 vs. Ag/AgCl;5 the energy of the LE state ELE is 2.34 eV, see the ESI†) is approximately +1.61 V. Without considering the Coulombic energy contribution due to a dipole character of the TICT state, it is evident that the intramolecular electron transfer between the separated LE state and a chalcogen substituent is feasible and increases in the order of the S < Se < Te substitution. However, in the presence of heavy atoms, such as Se and Te, formation of the TICT state must compete with efficient ISC (HAE), the probability of which increases in the same order. This explains counterintuitive findings in this work that the (TICT) fluorescence lifetime is not shortened with increasing atomic number of the substituents as observed for analogous 3-substituted BODIPYs20 (unfortunately, the scope of this study did not allow us to investigate why 3-chalcogeno-BODIPYs do not exhibit the TICT state), and that the most efficient ISC is observed for seleno-derivatives 3a and 7a and not for ditelluro-analogue 7d, in which the TICT fluorescence is even more efficient than ISC (radiationless transitions also largely account for excitation energy relaxation). Thus, the interplay of TICT formation and ISC in our BODIPY derivatives has an essential role in their spectroscopic properties and could be an interesting topic for further studies.
All BODIPY derivatives dissolved in acetonitrile were stable in the dark for months (ESI†) but slightly photolabile under irradiation with visible light. The quantum yields of photodecomposition (Φdecomp, λirr = 507 nm), determined using xanthene-9-carboxylic acid as an actinometer,5 were thus obtained to evaluate their photostability (Table 1; ESI†). In general, photodegradation (photo-deiodination) of 1 and 6 in acetonitrile was more efficient than decomposition of chalcogen-containing derivatives, in which irradiation led to degradation of the BODIPY core without apparent intermediacy of any species (Fig. S44 and S45, ESI†).
In summary, a facile synthetic method for the preparation of 2-chalcogen or 2,6-dichalogen-substituted BODIPYs, based on a Pd-catalyzed C–heteroatom Stille cross-coupling reaction using organostannanes, is introduced in this work. The procedure overcomes the limitations of aromatic nucleophilic substitution, and opens new ways for BODIPY core functionalization. The synthesized chalcogen derivatives exhibit interesting photophysical properties which are interpreted in terms of the interplay between the formation of an emissive TICT state, responsible for the observed large Stokes shifts, and intersystem crossing. The accessible synthesis and tunable properties of the novel BODIPY derivatives are important attributes for their future applications.
Support for this work was provided by the Czech Science Foundation (GA13-25775S). This work was supported by the Czech Ministry of Education, Youth and Sports (LO1214 and LM2015051).
References
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- G. Duran-Sampedro, I. Esnal, A. R. Agarrabeitia, J. Bañuelos Prieto, L. Cerdán, I. García-Moreno, A. Costela, I. Lopez-Arbeloa and M. J. Ortiz, Chem. – Eur. J., 2014, 20, 2646–2653 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- E. Palao, T. Slanina, L. Muchová, T. Šolomek, L. Vítek and P. Klán, J. Am. Chem. Soc., 2016, 138, 126–133 CrossRef CAS PubMed.
- N. Umeda, H. Takahashi, M. Kamiya, T. Ueno, T. Komatsu, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, ACS Chem. Biol., 2014, 9, 2242–2246 CrossRef CAS PubMed.
- N. Rubinstein, P. Liu, E. W. Miller and R. Weinstain, Chem. Commun., 2015, 51, 6369–6372 RSC.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577–6595 CrossRef CAS.
- L. Bonardi, G. Ulrich and R. Ziessel, Org. Lett., 2008, 10, 2183–2186 CrossRef CAS PubMed.
- L. Jiao, W. Pang, J. Zhou, Y. Wei, X. Mu, G. Bai and E. Hao, J. Org. Chem., 2011, 76, 9988–9996 CrossRef CAS PubMed.
- M. J. Ortiz, A. R. Agarrabeitia, G. Duran-Sampedro, J. B. Prieto, T. A. Lopez, W. A. Massad, H. A. Montejano, N. A. Garcia and I. L. Arbeloa, Tetrahedron, 2012, 68, 1153–1162 CrossRef CAS.
- C. F. A. Gómez-Durán, I. Esnal, I. Valois-Escamilla, A. Urías-Benavides, J. Bañuelos, I. López Arbeloa, I. García-Moreno and E. Peña-Cabrera, Chem. – Eur. J., 2016, 22, 1048–1061 CrossRef PubMed.
- G. Duran-Sampedro, A. R. Agarrabeitia, L. Cerdan, M. E. Perez-Ojeda, A. Costela, I. Garcia-Moreno, I. Esnal, J. Banuelos, I. L. Arbeloa and M. J. Ortiz, Adv. Funct. Mater., 2013, 23, 4195–4205 CrossRef CAS.
- V. Lakshmi, M. Rajeswara Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501–2517 CAS.
- T. Solomek, J. Wirz and P. Klan, Acc. Chem. Res., 2015, 48, 3064–3072 CrossRef CAS PubMed.
- S. Zhu, J. Bi, G. Vegesna, J. Zhang, F.-T. Luo, L. Valenzano and H. Liu, RSC Adv., 2013, 3, 4793–4800 RSC.
- M. Gupta, S. Mula, M. Tyagi, T. K. Ghanty, S. Murudkar, A. K. Ray and S. Chattopadhyay, Chem. – Eur. J., 2013, 19, 17766–17772 CrossRef CAS PubMed.
- T.-I. Kim, S. Park, Y. Choi and Y. Kim, Chem. – Asian J., 2011, 6, 1358–1361 CrossRef CAS PubMed.
- L. C. D. de Rezende, S. M. G. de Melo, S. Boodts, B. Verbelen, W. Dehaen and F. da Silva Emery, Org. Biomol. Chem., 2015, 13, 6031–6038 CAS.
- J. Al Anshori, T. Slanina, E. Palao and P. Klan, Photochem. Photobiol. Sci., 2016, 15, 250–259 CAS.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- W. Wu, X. Cui and J. Zhao, Chem. Commun., 2013, 49, 9009–9011 RSC.
- J. G. Knight, R. B. Alnoman and P. G. Waddell, Org. Biomol. Chem., 2015, 13, 3819–3829 CAS.
- G. Duran-Sampedro, E. Palao, A. R. Agarrabeitia, S. d. l. Moya, N. Boens and M. J. Ortiz, RSC Adv., 2014, 4, 19210–19213 RSC.
- Y. Nishiyama, K. Tokunaga and N. Sonoda, Org. Lett., 1999, 1, 1725–1727 CrossRef CAS.
- I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, J. Organomet. Chem., 2000, 605, 96–101 CrossRef CAS.
- J. X. Chen and G. T. Crisp, Synth. Commun., 1992, 22, 683–686 CrossRef CAS.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- H. J. Reich, J. M. Renga and I. L. Reich, J. Am. Chem. Soc., 1975, 97, 5434–5447 CrossRef CAS.
- R. Rossi, F. Bellina and L. Mannina, Tetrahedron, 1997, 53, 1025–1044 CrossRef CAS.
- D. N. Harpp, T. Aida and T. H. Chan, Tetrahedron Lett., 1979, 20, 2853–2856 CrossRef.
- E. Palao, G. Duran-Sampedro, S. de la Moya, M. Madrid, C. García-López, A. R. Agarrabeitia, B. Verbelen, W. Dehaen, N. Boens and M. J. Ortiz, J. Org. Chem., 2016, 81, 3700–3710 CrossRef CAS PubMed.
- C. H. Schiesser and M. A. Skidmore, J. Org. Chem., 1998, 63, 5713–5715 CrossRef CAS.
- R. Hu, E. Lager, A. l. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- X.-F. Zhang, X. Yang, K. Niu and H. Geng, J. Photochem. Photobiol., A, 2014, 285, 16–20 CrossRef CAS.
- R. P. Sabatini, T. M. McCormick, T. Lazarides, K. C. Wilson, R. Eisenberg and D. W. McCamant, J. Phys. Chem. Lett., 2011, 2, 223–227 CrossRef CAS.
- J. Bañuelos, Chem. Rec., 2016, 16, 335–348 CrossRef PubMed.
- Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192–2206 CrossRef CAS PubMed.
- J.-L. Jin, H.-B. Li, Y. Geng, Y. Wu, Y.-A. Duan and Z.-M. Su, ChemPhysChem, 2012, 13, 3714–3722 CrossRef CAS PubMed.
- A. Y. Bochkov, I. O. Akchurin, O. A. Dyachenko and V. F. Traven, Chem. Commun., 2013, 49, 11653–11655 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- E. L. Mertz, V. A. Tikhomirov and L. I. Krishtalik, J. Phys. Chem. A, 1997, 101, 3433–3442 CrossRef CAS.
- P. Horváth, P. Šebej, T. Šolomek and P. Klán, J. Org. Chem., 2015, 80, 1299–1311 CrossRef PubMed.
- L.-S. Choi, Chem. Commun., 1998, 893–894 RSC.
- M. Viard, J. Gallay, M. Vincent, O. Meyer, B. Robert and M. Paternostre, Biophys. J., 1997, 73, 2221–2234 CrossRef CAS PubMed.
- J. B. Kelber, N. A. Panjwani, D. Wu, R. Gomez-Bombarelli, B. W. Lovett, J. J. L. Morton and H. L. Anderson, Chem. Sci., 2015, 6, 6468–6481 RSC.
- S. S. Palayangoda, X. Cai, R. M. Adhikari and D. C. Neckers, Org. Lett., 2008, 10, 281–284 CrossRef CAS PubMed.
- L. Ludvikova, P. Fris, D. Heger, P. Sebej, J. Wirz and P. Klan, Phys. Chem. Chem. Phys., 2016, 18, 16266–16273 RSC.
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 5533–5539 CrossRef CAS PubMed.
- Z. R. Grabowski and J. Dobkowski, Pure Appl. Chem., 1983, 55, 245–252 CrossRef CAS.
- L. Engman, J. Persson, C. M. Andersson and M. Berglund, J. Chem. Soc., Perkin Trans. 2, 1992, 1309–1313 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; spectroscopy; synthesis and compound characterization; and NMR and HRMS and optical spectra. See DOI: 10.1039/c6cc06923a |
| This journal is © The Royal Society of Chemistry 2016 |