
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triple-decker sandwich complexes with a bent cyclo-P5 middle-deck†
Eric
Mädl
a,
Eugenia
Peresypkina
bc,
Alexey Y.
Timoshkin
 d and
Manfred
Scheer
*a
d and
Manfred
Scheer
*a
aInstitut für Anorganische Chemie, Universität Regensburg, 93051 Regensburg, Germany. E-mail: Manfred.scheer@ur.de; Web: http://www.ur.de/chemie-pharmazie/anorganische-chemie-scheer
bNikolaev Institute of Inorganic Chemistry, Siberian Division of RAS, Acad. Lavrentyev str. 3, 630090 Novosibirsk, Russia
cNovosibirsk State University, Pirogava str. 2, 630090 Novosibirsk, Russia
dSt. Petersburg State University, University pr. 26, 198504 Old Peterhoff, St. Petersburg, Russia
First published on 15th September 2016
Abstract
New types of triple-decker complexes with an organo-substituted P5 middle-deck were synthesized by the reaction of [Cp*Fe(η4-P5R)]− (1a: R = CH2SiMe3; 1b: R = NMe2) with halogeno-bridged transition metal dimers [Cp′′′MX]2 (M = Cr, Fe, Co, Ni; X = Cl, Br). By oxidation of [(Cp*Fe)(Cp′′′Co)(μ,η4:3-P5CH2SiMe3)] 2a with [Cp2Fe][PF6], the cationic complex [(Cp*Fe)(Cp′′′Co)(μ,η5:4-P5CH2SiMe3)]+ was isolated. The electronic structure of the synthesized complexes was elucidated by DFT calculations.
Ferrocene is one of the most frequently used organometallic reagents in chemistry, with very broad applications.1–3 This 65 year old molecule4,5 does not only show a fascinating chemistry in redox processes but particularly as a starting material for subsequent reactions. Its ability to be metalated6,7 or to undergo Friedel–Craft reactions8,9 renders it a valuable starting material in organometallic synthesis. The isolobal analogue of ferrocene is pentaphosphaferrocene and its Cp* derivative [Cp*Fe(η5-P5)] (I) was first discovered in 1987.10 The majority of reactivity studies of I is dedicated to the coordination chemistry towards Lewis acidic coordination moieties, forming 1D and 2D coordination polymers11–15 or spherical supramolecular clusters.16–20 In cothermolysis or cophotolysis reactions with organometallic reagents, fragmentations and deformations of the cyclo-P5 ring of I occur.21–24 A new direction for the reactivity of pentaphosphaferrocene opened up when I was used in redox processes25–27 and especially when it was converted by nucleophiles.28 In the latter case, a selective functionalization of the P5 ring in I was achieved. As a result, monoanionic complexes of the type [Cp*Fe(η4-P5R)]− (R = CH2SiMe3, NMe2, PH2) were isolated, leading to new perspectives in the chemistry of I. These monoanionic complexes raised the question, whether reacting them with electrophiles leads to a reformation of the initial cyclo-P5 ring (by the retention of the former substitution), or if a rearrangement takes place to give products with novel structures. Moreover, so far only few triple-decker complexes exhibiting a cyclo-P5 middle deck are known. Starting from P4 in thermolysis reactions, the compounds [(CpBIGMn)2(μ,η5:5-P5)] (CpBIG = C5(4-nBuC6H4)5) and [(CpRCr)2(μ,η5:5-P5)] (CpR = Cp, Cp*) are obtained.29–31 Starting from I, some cationic triple-decker compounds [(Cp*M)(CpRM′)(η5:5-P5)]+ (M, M′ = Fe, Ru; CpR = Cp, Cp*), containing group 8 elements, have been reported.32,33 Furthermore, mixed-metal lanthanide-iron compounds with a cyclo-P5 middle-deck26 and triple-decker complexes consisting of I and a [M(CO)3] fragment (M = Cr, Mo, W) are known.34 Herein, we report the synthesis of the first neutral complexes with a functionalized P5 middle-deck under mild conditions. By using different transition metal halides [Cp′′′MX]2 (M = Cr, Fe, Co, Ni; X = Cl, Br), a broad variety of different triple-decker complexes are easily accessible. Their bonding situation has been investigated by quantum chemical computations.
The reaction of 1a/1b with the transition metal dimers [Cp′′′MX]2 (M = Cr, Fe, Co, Ni; X = Cl, Br) leads to the triple-decker complexes 2–5, containing the whole 3d element series from Cp′′′Cr to Cp′′′Ni decks (Scheme 1). Unfortunately, despite several efforts, we were not able to synthesize the missing manganese containing triple-decker complex in this series.35 Whereas the compounds 3–5 are diamagnetic, the obtained cobalt/iron triple-decker 2a/2b are paramagnetic. The EPR spectra of 2a and 2b in toluene at 77 K show an isotropic signal for both compounds, centred at giso = 2.069 and 2.076, respectively, with no hyperfine coupling. The determination of the effective magnetic moment of 2a and 2b in solution by the Evans method results in one unpaired electron (2a: μeff = 2.14 μB; 2b: μeff = 1.83 μB). As DFT calculations on the B3LYP/def2-SVP level of theory show, the single-occupied molecular orbitals (SOMO) of 2a and 2b are very similar, indicating the minor influence of the substituent of the cyclo-P5 ring on the features of the SOMO (Fig. 1).36 All SOMOs are delocalized, but the analysis of the spin density reveals that the metal centre bonded to the Cp′′′ ligand exhibits the highest spin density. The calculated atomic spin densities of 2a/2b show that the Co atom possesses the highest positive spin density (about 62%), followed by the Fe atom (about 19%).
 | ||
| Scheme 1 Reactions of I: (i) LiCH2SiMe3 in Et2O or LiNMe2 in THF, r.t.; reaction of 1a: (ii) [Cp′′′CoCl]2 in THF, r.t., (iv) [Cp′′′CrCl]2 in THF, r.t.; reaction of 1b: (ii) [Cp′′′CoCl]2 or [Cp′′′NiBr]2 in THF, r.t.; (iii) [Cp′′′FeBr]2 in THF, r.t.; reaction of 2a: (v) [Cp2Fe][PF6] in THF, r.t. −50 °C → r.t. Yields in parentheses. | ||
 | ||
| Fig. 1 Left: Isosurface of the single-occupied molecular orbitals (SOMO) in 2a calculated on the B3LYP/def2-SVP level of theory. Hydrogens are omitted for clarity. Right: Cyclic voltammogram of 2a recorded at a platinum disc electrode in CH2Cl2 at 100 mV s−1 and referenced against fc/fc+; supporting electrolyte [Bu4N][PF6] (0.1 mol L−1). | ||
The 31P NMR spectrum of the diamagnetic nickel/iron triple-decker complex 3 shows an AXX′ZZ′ spin system, with one triplet of triplets centered at 40.6 ppm and two multiplets centered at −30.29 and −53.9 ppm. For the iron/iron triple-decker complex 4, the 1H NMR spectrum shows only sharp signals. However, in contrast to the triple-decker complex 3, in the 31P NMR spectrum of 4 one sharp signal at 73.8 ppm, one broad signal at −131.1 ppm and one very broad signal at −150.8 ppm are observed at room temperature. By cooling down the sample to 193 K, five broad signals in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1:1 integral ratio are monitored in the 31P NMR spectrum, centered at 65.8, 42.2, −104.5, −195.0 and −344.9 ppm, revealing a dynamic behavior of the P5 ring. In this process, the phosphorus atoms adjacent to the substituted P atom coordinate alternatingly to the Cp*Fe fragment, and the Cp′′′Fe fragment slips over the middle-deck. At 193 K, the signals of the Cp′′′ ligand in the 1H NMR spectrum of 4 become very broad, showing that the free rotation of the cyclopentadienyl ligand is slowed down.
:1:1:1:1 integral ratio are monitored in the 31P NMR spectrum, centered at 65.8, 42.2, −104.5, −195.0 and −344.9 ppm, revealing a dynamic behavior of the P5 ring. In this process, the phosphorus atoms adjacent to the substituted P atom coordinate alternatingly to the Cp*Fe fragment, and the Cp′′′Fe fragment slips over the middle-deck. At 193 K, the signals of the Cp′′′ ligand in the 1H NMR spectrum of 4 become very broad, showing that the free rotation of the cyclopentadienyl ligand is slowed down.
When 1a is reacted with [Cp′′′CrCl]2, the 31P NMR spectrum of the reaction solution shows a broad doublet of doublets at 281.1, one broad doublet at 9.5 ppm and a triplet of triplets at −66.7 ppm for the chromium/iron triple-decker complex 5. Also, one set of signals of an unidentified byproduct is observed (in about 13%), but 5 can be isolated and purified further by recrystallization. By cooling down the sample of 5 in the 31P NMR spectrum at 253 K, the signals become sharp and a fine structure is determined.36 The simulation of this spectrum reveals unusual coupling constants: the 1JP–P coupling (P2–P3) between the P2 dumbbell and the P3 allylic moiety is remarkably small (22.65 Hz). This is consistent with the corresponding elongated P–P distance (vide infra). The 2JP–P coupling (P1–P3) is comparably large with a value of about 100 Hz. Usually the absolute value of a 1JP–P coupling is significantly higher compared to a 2JP–P coupling, as it is observed in 3 or the starting materials 1a/1b. This unusual behaviour may originate from the orbital interaction between the phosphorus atoms via the metal centres (as it is seen in the HOMO−3,36 which has contributions from the atomic orbitals of the P1, Cr and Fe atom, respectively, and the orbital of the P3–P4 unit).
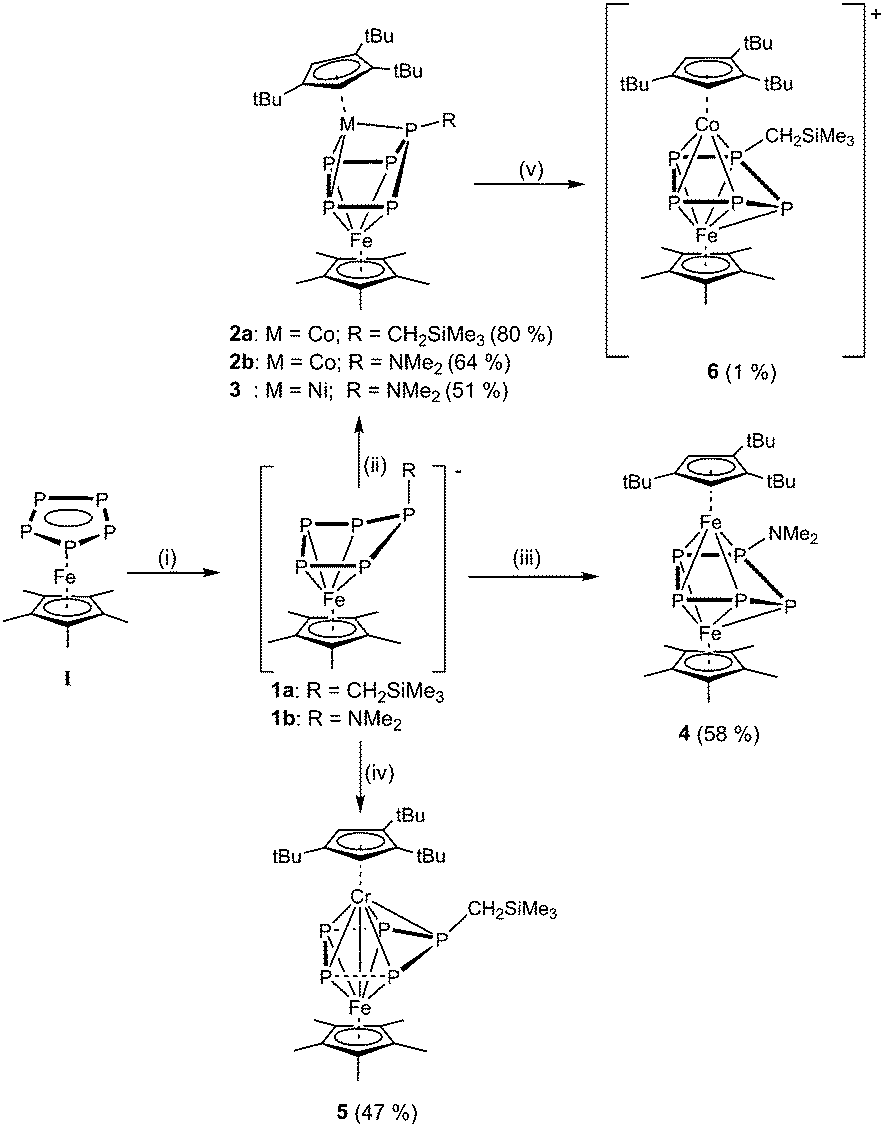
Compounds 2–5 could all be characterized by X-ray structure analysis. The triple-decker complexes 2a, 2b and 3 exhibit a similar structural motif, in which the P5 ligand adopts a η4:η3-coordination mode (Fig. 2). Compared to 1a/1b, in 2a/2b and 3 the enveloped conformation of the P5 unit becomes more distinct, with three of the five phosphorus atoms (P1, P3, P4; labeling according to Fig. 2) coordinating to the Cp′′′ metal fragment. The phosphorus atom, which does not lie in the η4-P4 plane, still bears the organic rest. In 1a/1b, all P–P bonds of the P5 ring exhibit double bond character. In the triple-decker sandwich complex 2a the P–P bond lengths range from 2.1771(7) Å to 2.3530(7) Å. Compounds 2b and 3 show similar values (2b: 2.1569(8)–2.4132(8) Å; 3: 2.1620(7)–2.4739(7) Å). Particularly noticeable is the rather long P3–P4 bond in the backbone of the P5 ring of 2a, 2b and 3 (in 2a: 2.3530(7) Å; 2b: 2.4132(8) Å; 3: 2.4738(7) Å). The corresponding Wiberg bond index (WBI) values for these bonds are 0.70, 0.67 and 0.51 for 2a, 2b, and 3, respectively.
 | ||
| Fig. 2 Molecular structure of 2a (left) and 3 (right). Ellipsoids are drawn at 50% probability level. H atoms bonded to carbon are omitted for clarity. | ||
The elongation of this P–P bond, by going from 2a to 3, is in line with the increased electron density in the P5 moiety (compound 2a features a CH2SiMe3 group exhibiting a +I effect; compound 2b features an NMe2 group exhibiting +M effect; compound 3 contains one additional electron due to the exchange of the cobalt with a nickel atom). Furthermore in 2b and 3, the NMe2 group is planarly arranged with the sum of the angles around the nitrogen atom of 360° (in 2a: 359.99°; in 3: 359.96°), and the N–P bond possesses double bond character (2b: 1.665(2); 3: 1.671(2) Å), reflecting the donating character of the NMe2 group in these compounds. The WBI values for the Co1–P1 bond order in 2b (0.76) are similar to 2a (0.75) upon introduction of the more electronegative NMe2 substituent on the P1 atom. The Mulliken charges in 2a and 2b are akin.36
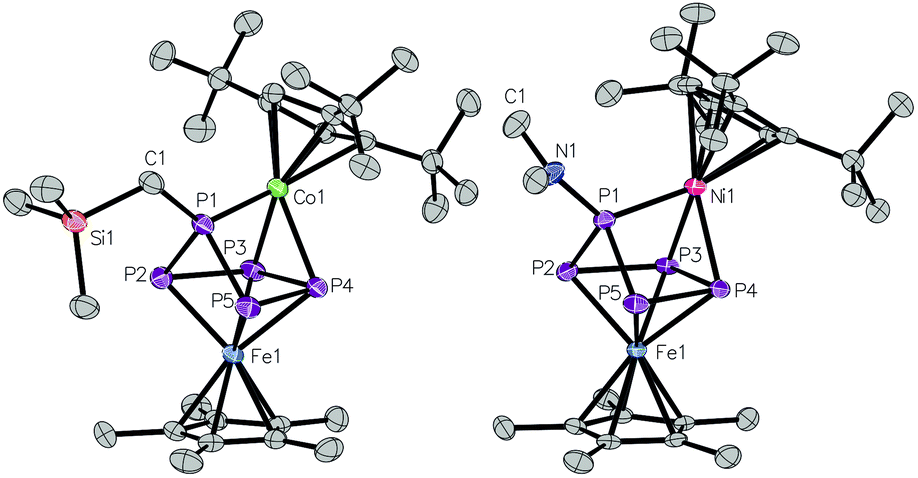
By comparing 2a, 2b and 3 with the iron/iron complex 4, the conformation of the P5 ring changes (Fig. 3). Due to a distortion of the phosphorus ring, the Fe2–P5 bond (2.6313(5) Å) is prolonged (remaining Fe–P bond lengths in 4: 2.1362(5)–2.4690(5) Å, Fig. 3), thus the coordination mode of the P5 ligand is best described as η4:η4. Furthermore, the enveloped P5 ring of 4 is bent towards the Cp*Fe fragment – instead towards the Cp′′′M fragment, as observed in the triple-decker sandwich complexes 2a,b. The phosphorus atom, which bears the dimethylamine rest, lies within a distorted η4-P4 plane. In comparison to 2a, 2b and 3, the P2–P3 (2.3587(6) Å) and P4–P5 (2.3187(6) Å) bonds are considerably longer in 4 and the P3–P4 bond exhibits double bond character in 4 (2.1054(6) Å).
 | ||
| Fig. 3 Molecular structure of 4 (left) and 5 (right). Ellipsoids are drawn at 50% probability level. H atoms bonded to carbon are omitted for clarity. | ||
If the P5 moiety coordinates to the electron deficient Cp′′′Cr metal fragment in 5, a significant structural change of the initial cyclo-P5 ring is found. The original cylco-P5 ring of 5 is broken into a P2 dumbbell (P3–P4: 2.1040(7) Å; labeling according to Fig. 3) and a P3 fragment (P1–P5: 2.1549(6) Å; P1–P2: 2.1558(7) Å), while a Fe–Cr bond (2.6252(4) Å) is formed. The small WBI of the P2–P3 and P4–P5 distances (0.22 each) also reflect the P2/P3 separation of the former P5 ring. Compared to the Cr–Fe bond length in [CpFe(CO)2Cr(CO)3Cp] (2.901(1) Å)37 or in [(CpCr)(CpFe)(η4:4-oct)] (oct = cyclooctatetraene), (2.7261(8) Å),38 the metal–metal bond in 5 is shortened. The WBI for the Cr–Fe bond equals 0.47, which is considerably larger in comparison to the M–M WBI of the previously discussed triple-decker compounds 2a–3 (0.12–0.13).
The iron/nickel and iron/iron containing complexes 3 and 4 are stable and formally only differ by one electron in comparison to the iron/cobalt containing triple-decker complexes 2a/2b. Therefore, the electrochemical properties of 2a/2b were investigated. The cyclic voltammogram of 2a in CH2Cl2 shows two oxidations and one reduction (Fig. 1). The first oxidation occurs at a half potential of −0.79 V and exhibits a reversible character (ip(reverse)/ip(forward) = 0.82).39 The second oxidation at 0.39 V is considered irreversible. At −1.67 V, a reversible reduction is observed (ip(reverse)/ip(forward) = 0.98). The cyclic voltammogram of 2b exhibits similar features,36 with one reversible oxidation at −0.88 V (ip(reverse)/ip(forward) = 0.98) and a following irreversible one at 0.39 V.39 A reversible reduction is observed at −1.61 V (ip(reverse)/ip(forward) = 0.97).
Based on these studies, we chose the oxidizing agents [Cp2Fe][PF6] for the chemical oxidization of 2a, which has a half potential of −0.59 V against fc/fc+ in MeCN.40 Contrary to the expectation that [(Cp*Fe)(Cp′′′Co)(μ,η4:3-P5CH2SiMe3)]+ (6) should be diamagnetic in analogy to 4, in the 31P NMR spectrum of the reaction solution of 2a with [Cp2Fe][PF6] only signals of low intensity for some minor impurities could be determined, as well as a septet centered at −140.4 ppm for the [PF6]− ion. DFT calculations show that the triplet state of the cation of [6]+ in the gas phase is by 16.7 kJ mol−1 lower in energy than the singlet state. A few single crystals of [6][PF6] were obtained from a Et2O solution (Scheme 1). The X-ray structure analysis reveals that [6]+ is not just isoelectronical to the triple-decker complex [(Cp*Fe)(Cp′′′Fe)(μ,η4:4-P5NMe2)] (4), but that 2a undergoes a structural rearrangement during the oxidation, resulting in [6]+ to be isostructural to 4 (Fig. 4). The phosphorus atom, which bears the organic rest and was out of the η4-P4 plane in 2a, interchanges hereby the position with an unsubstituted phosphorus atom from the η4-P4 plane. Unfortunately, we did not succeed in isolating any reduced products of 2a or 2b, by using K or KH as reducing agents despite many attempts.
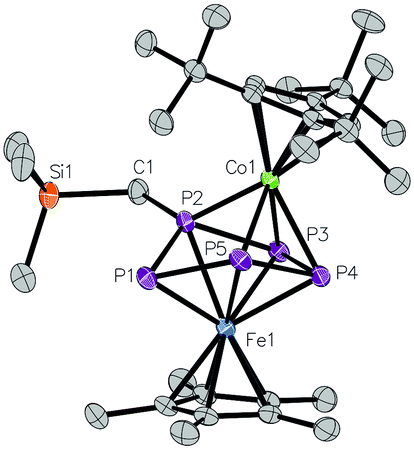 | ||
| Fig. 4 Molecular structure of cationic part of [6][PF6]. Ellipsoids are drawn at 50% probability level. H atoms bonded to carbon are omitted for clarity. | ||
In summary, we showed a subsequent chemistry of the anionic functionalized pentaphosphaferrocenes, by reacting them with transition metal halide dimers. That way, several unique neutral triple-decker sandwich complexes with unprecedented functionalized cyclo-P5 middle-decks were obtained. The integrity of the initial cyclo-P5 middle-deck depends strongly on the electronic situation of the coordinating metal fragments, which leads from a structural rearrangement of the enveloped P5 moiety in 2a/2b, 3 and 4 to a complete fragmentation of the P5 ring, as seen for [(Cp*Fe)(Cp′′′Cr)(μ,η4:5-P5CH2SiMe3)] (5). In addition, the triple-decker complexes 2a and 2b show interesting electrochemical properties and a change of conformation of the P5 moiety is observed upon oxidation. The successful salt elimination of the anionic pentaphosphaferrocene derivatives opens new avenues for the chemistry of pentaphosphaferrocene. Further functionalization of the P5 ring should now be possible, which will lead to transfer reactions of the P5 moiety or to the isolation of uncoordinated organo-substituted phosphorus derivatives.
The authors thank the Deutsche Forschungsgemeinschaft for funding and Dr M. Walter (Technical University of Braunschweig) for providing [Cp′′′MnI]2.
Notes and references
- C. Ornelas, New J. Chem., 2011, 35, 1973–1985 RSC.
- R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674–7715 CrossRef PubMed.
- X. Zhai, H. Yu, L. Wang, Z. Deng, Z.-U. Abdin, R. Tong, X. Yang, Y. Chen and M. Saleem, Appl. Organomet. Chem., 2016, 30, 62–72 CrossRef CAS.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- S. A. Miller, J. A. Tebboth and J. F. Tremaine, J. Chem. Soc., 1952, 632–635 RSC.
- M. Rausch, M. Vogel and H. Rosenberg, J. Org. Chem., 1957, 22, 900–903 CrossRef CAS.
- R. A. Benkeser, D. Goggin and G. Schroll, J. Am. Chem. Soc., 1954, 76, 4025 CrossRef CAS.
- G. P. Sollott and E. Howard, J. Org. Chem., 1962, 27, 4034–4040 CrossRef CAS.
- G. D. Broadhead, J. M. Osgerby and P. L. Pauson, J. Chem. Soc., 1958, 650–656 RSC.
- O. J. Scherer and T. Brück, Angew. Chem., Int. Ed., 1987, 26, 59 Search PubMed.
- O. J. Scherer, J. Vondung and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1989, 28, 1355–1357 CrossRef.
- M. Fleischmann, S. Welsch, H. Krauss, M. Schmidt, M. Bodensteiner, E. V. Peresypkina, M. Sierka, C. Gröger and M. Scheer, Chem. – Eur. J., 2014, 20, 3759–3768 CrossRef CAS PubMed.
- S. Welsch, L. J. Gregoriades, M. Sierka, M. Zabel, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2007, 46, 9323–9326 CrossRef CAS PubMed.
- M. Scheer, L. J. Gregoriades, A. V. Virovets, W. Kunz, R. Neueder and I. Krossing, Angew. Chem., Int. Ed., 2006, 45, 5689–5693 CrossRef CAS PubMed.
- J. Bai, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 1737–1740 CrossRef CAS.
- J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed.
- M. Scheer, J. Bai, B. P. Johnson, R. Merkle, A. V. Virovets and C. E. Anson, Eur. J. Inorg. Chem., 2005, 4023–4026 CrossRef CAS.
- M. Scheer, A. Schindler, J. Bai, B. P. Johnson, R. Merkle, R. Winter, A. V. Virovets, E. V. Peresypkina, V. A. Blatov, M. Sierka and H. Eckert, Chem. – Eur. J., 2010, 16, 2092–2107 CrossRef CAS PubMed.
- M. Scheer, A. Schindler, C. Gröger, A. V. Virovets and E. V. Peresypkina, Angew. Chem., Int. Ed., 2009, 48, 5046–5049 CrossRef CAS PubMed.
- M. Scheer, A. Schindler, R. Merkle, B. P. Johnson, M. Linseis, R. Winter, C. E. Anson and A. V. Virovets, J. Am. Chem. Soc., 2007, 129, 13386–13387 CrossRef CAS PubMed.
- M. Detzel, T. Mohr, O. J. Scherer and G. Wolmershäuser, Angew. Chem., 1994, 106, 1142–1144 CrossRef CAS.
- O. J. Scherer, T. Mohr and G. Wolmershäuser, J. Organomet. Chem., 1997, 529, 379–385 CrossRef CAS.
- B. Koch, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 2000, 626, 1797–1802 CrossRef CAS.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972–2976 CrossRef CAS PubMed.
- T. Li, J. Wiecko, N. A. Pushkarevsky, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 9491–9495 CrossRef CAS PubMed.
- T. Li, M. T. Gamer, M. Scheer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2013, 49, 2183–2185 RSC.
- E. Mädl, M. V. Butovskii, G. Balázs, E. V. Peresypkina, A. V. Virovets, M. Seidl and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7643–7646 CrossRef PubMed.
- S. Heinl, G. Balazs, M. Bodensteiner and M. Scheer, Dalton Trans., 2016, 45, 1962–1966 RSC.
- O. J. Scherer, J. Schwalb, G. Wolmershäuser, W. Kaim and R. Gross, Angew. Chem., Int. Ed., 1986, 25, 363–364 CrossRef.
- L. Y. Goh, R. C. S. Wong, C. K. Chu and T. W. Hambley, J. Chem. Soc., Dalton Trans., 1990, 977–982 RSC.
- Alexander R. Kudinov, Dmitry A. Loginov, Zoya A. Starikova, Pavel V. Petrovskii, M. Corsini and P. Zanello, Eur. J. Inorg. Chem., 2002, 3018–3027 CrossRef CAS.
- D. A. Loginov, I. D. Baravi, O. I. Artyushin, Z. A. Starikova, P. V. Petrovskii and A. R. Kudinov, Russ. Chem. Bull., 2010, 59, 1312–1316 CrossRef CAS.
- B. Rink, O. J. Scherer, G. Heckmann and G. Wolmershauser, Chem. Ber., 1992, 125, 1011–1016 CrossRef CAS.
- Although a reaction of 2a with [Cp′′′MnI]2 is observed (no signals of 2a or any other compounds are monitored in the 31P NMR spectrum of the reaction mixture), no products could be isolated, even after chromatographic work-up.
- cf. ESI†.
- W. A. Herrmann, J. Rohrmann, E. Herdtweck, C. Hecht, M. L. Ziegler and O. Serhadli, J. Organomet. Chem., 1986, 314, 295–305 CrossRef CAS.
- J. Heck, P. Maurice, J. A. Hermans, A. B. Scholten, W. P. J. H. Bosman, G. Meyer, T. Staffel, R. Stürmer and M. Wünsch, Z. Anorg. Allg. Chem., 1992, 611, 35–42 CrossRef CAS.
- Referenced against ferrocene.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterisation and details of the DFT calculations. CCDC 1499116–1499121. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06883a |
| This journal is © The Royal Society of Chemistry 2016 |