
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRatiometric and photoconvertible fluorescent protein-based voltage indicator prototypes†
A. S.
Abdelfattah
a,
V.
Rancic
b,
B.
Rawal
b,
K.
Ballanyi
b and
R. E.
Campbell
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: robert.e.campbell@ualberta.ca
bDepartment of Physiology, University of Alberta, Edmonton, Alberta T6G 2H7, Canada
First published on 14th November 2016
Abstract
To expand the toolbox of fluorescent protein-based voltage indicators, we explored two distinct protein design strategies. Using these design strategies, we created three new voltage indicators: a red intensiometric voltage indicator (tdFlicR Δ110AR), a green/red ratiometric voltage indicator (tdFlicR-VK-ASAP), and a green to red photoconvertible voltage indicator (FlicGR1).
At its most fundamental level, brain function emerges from the complex circuitry of neurons receiving synaptic inputs and firing action potentials. Analysis of the propagation of signals through neuronal circuits can be achieved by measurement of changes in membrane potential of interacting neurons using synthetic dye-based voltage indicators and genetically encoded voltage indicators (GEVIs).1 Compared to synthetic dye-based indicators, GEVIs provide cell-type specific labeling of a genetically-defined subset of cells, making GEVIs more suitable for studying neuronal circuitry.
Protein engineers have used several protein design strategies to couple membrane potential changes to fluorescence changes of genetically encoded fluorescent domains. One family of GEVIs relies on fusion of a voltage sensitive domain (VSD) to one fluorescent protein (FP) or to a Förster resonance energy transfer (FRET) pair of FPs. Voltage-dependent conformational changes of the VSD modulates fluorescence of one attached FP, or FRET efficiency between two FPs. Prototypes have been engineered using various VSDs,2 however, VSDs from voltage sensitive phosphatases3 of the marine organism Ciona intestinalis (CiVSD)4 and from chicken Gallus gallus (GgVSD)5 are most efficiently targeted to the membrane of mammalian cells.
We recently reported the development of a red FP-based voltage indicator (FlicR1) that is based on CiVSD and a circularly permutated (cp) red FP.4h We now report the use of FlicR1 as a template to explore new designs that expand the toolbox of FP-based GEVIs. Specifically, we report the development of voltage indicators that use the FlicR backbone and two copies of VSDs linked in tandem by a cpFP. This design allowed us to create two new GEVIs: a red intensiometric voltage indicator (tdFlicR Δ110AR) and a green/red ratiometric voltage indicator (tdFlicR-VK-ASAP). In addition, we describe the development of a photoconvertible voltage indicator, designated as FlicGR1 (fluorescent indicator for voltage imaging – green to red), which can be photoconverted from a green to a red fluorescent form using violet light illumination. FlicGR1 was engineered by replacing the red FP of FlicR1 with a photoconvertible FP.
Crystal structures of CiVSD in both the active and resting conformations were recently reported.6 Based on these atomic structures, it is apparent that CiVSD forms a homodimer through the S1/S4 interface. This insight prompted us to design single-polypeptide voltage indicators with tandem VSDs that could preserve the VSD dimer interface intramolecularly. This design contrasts with the current crop of FP-based voltage indicators that utilize a single VSD domain (Fig. S1, ESI†). We reasoned that a tandem VSD dimer would undergo voltage-dependent conformational changes that could potentially be harnessed to induce substantial changes in the cpFP chromophore environment. In addition, a second VSD might enable us to engineer a two-color ratiometric voltage indicator by harnessing the movement of both VSDs to modulate the fluorescence of two different colors of FPs.
To develop a red fluorescent voltage indicator based on a tandem VSD dimer, we fused a second CiVSD to the C-terminus of FlicR1 (Fig. S1F, ESI†). Simple fusion led to a construct with dim fluorescence. We reasoned that the protein sequence at the fusion site would likely have a strong effect on protein folding and maturation. Inspection of the X-ray crystal structure revealed that the N-terminal 105 residues of CiVSD are sufficiently disordered as to not be visible in the crystal structure.6 Furthermore, previous studies have demonstrated that the N-terminal 70 residues of CiVSD are not necessary for protein folding or membrane targeting.4a To identify the optimal length and composition of the linker at the fusion site between the cpmApple domain and the second CiVSD, we created a series of linker libraries by systematically truncating the unstructured region (Fig. 1A).
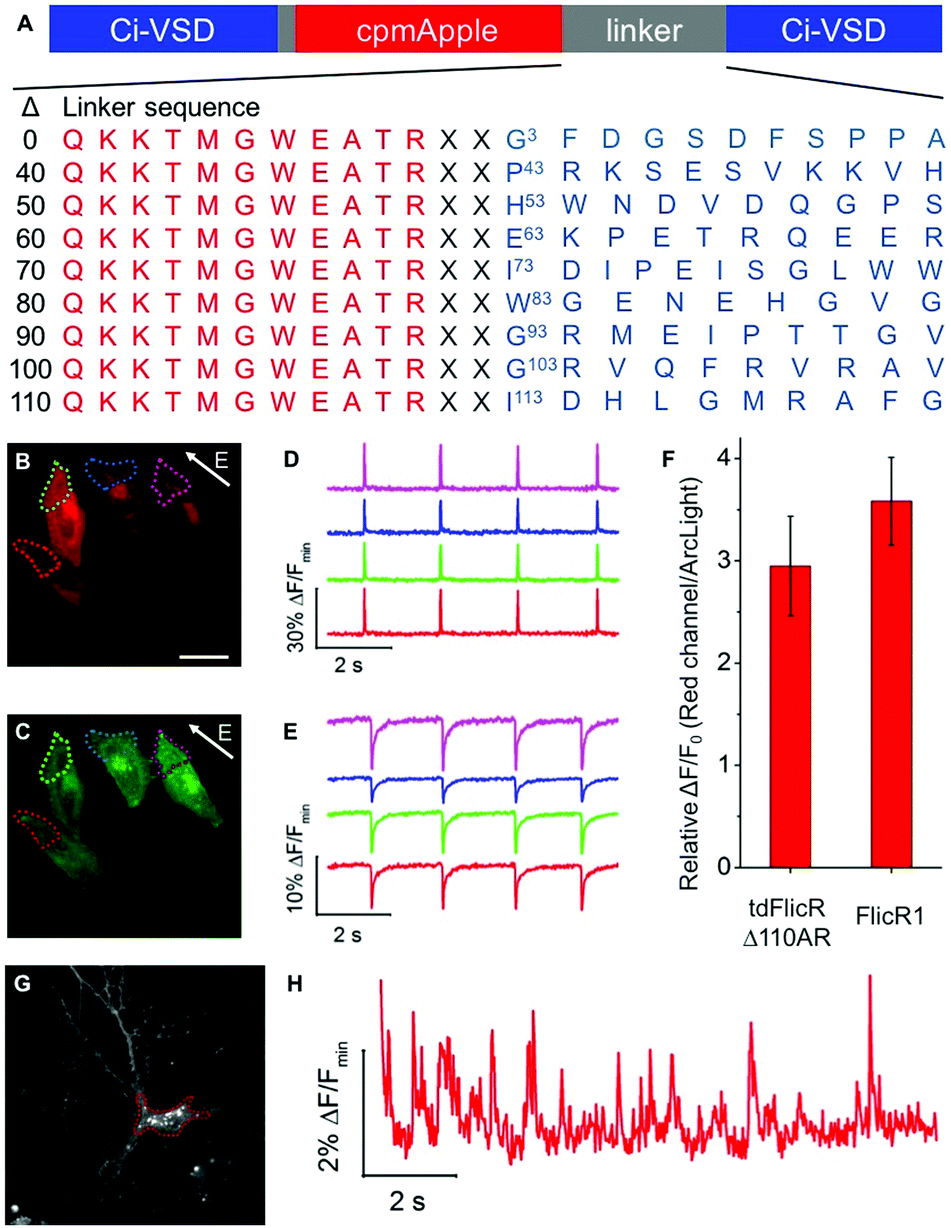 | ||
| Fig. 1 (A) Representation of tdFlicR linker libraries. X denotes a randomized residue. (B and C) Image of HeLa cells co-expressing tdFlicR Δ110AR (B) and ArcLight Q239 (C) (scale bar 10 μm). (D) tdFlicR Δ110AR fluorescence response of four regions, color-coded as indicated in (B), to ITV (10 ms, 25 V). (E) ArcLight Q239 fluorescence response of the same four regions indicated in (C) to ITV (10 ms, 25 V). (F) Comparison of voltage sensitivity between tdFlicR Δ110AR (n = 10 cells) and FlicR1 (n = 12 cells) represented as a ratio of response amplitude in HeLa cells compared to ArcLight Q239. Error bars are standard deviation. (G) Image of cultured hippocampal neuron expressing tdFlicR Δ110AR. (H) tdFlicR Δ110AR fluorescence response to spontaneous action potentials in the hippocampal neuron shown in (G). | ||
Linker libraries were screened for variants with high brightness, efficient membrane trafficking, and high voltage sensitivity, as schematically represented in Fig. S2 and described in the ESI.† To facilitate screening, we used a single plasmid that allows expression in both E. coli and mammalian cells (see ESI†).4h Arclight Q239 (a green voltage indicator)4e was used as an internal reference to assess membrane trafficking and voltage sensitivity (Fig. S2, ESI†).
We identified a promising variant from the Δ110 library with Ala and Arg at the fusion site (designated tdFlicR Δ110AR) for further characterization in HeLa cells and cultured neurons. tdFlicR Δ110AR localized to the plasma membrane of HeLa cells (Fig. 1B) and gave robust fluorescence changes in response to induced transmembrane voltage (ITV) (Fig. 1B–E). Relative to ArcLight Q239, tdFlicR Δ110AR has a 2.9 ± 0.5-fold (n = 9 HeLa cells) greater fluorescence response to a 10 ms ITV (Fig. 1F). To test the response of tdFlicR Δ110AR to spontaneous action potentials, we expressed the gene under the control of human synapsin I promoter in hippocampal neurons. Fluorescence imaging revealed that tdFlicR Δ110AR localized to the plasma membrane both at the cell body and in individual dendrites, but also formed intracellular puncta in the cell bodies similar to FlicR1 and other cpmApple-derived indicators (Fig. 1G).4h,7 When imaged at 100 Hz tdFlicR Δ110AR reported spontaneous activity of the transfected neurons with ∼2% ΔF/F0 increase in fluorescence (Fig. 1H).
Although we were unable to identify tdFlicR variants that had clear advantages compared to FlicR1, we recognized that the tdFlicR topology included a second VSD that could potentially be used to modulate the fluorescence of a second color of FP. This would enable us to construct a two-color ratiometric voltage indicator if the second FP was spectrally distinct from mApple and exhibited a decrease in fluorescence upon membrane depolarization. One GEVI that exhibits a decrease in green fluorescence in response to membrane depolarization is ASAP1,5 (Fig. S1C and S3, ESI†) making it a suitable scaffold for use in a two-color ratiometric voltage indicator using the tdFlicR design.
To develop a two-color ratiometric voltage indicator, we substituted the second CiVSD in tdFlicR variants with ASAP1 (Fig. 2A). We constructed a library of 400 protein variants in E. coli by varying two amino acids at the junction between cpmApple and ASAP1 and selected colonies that exhibit bright green and red fluorescence. Next we transfected mammalian cells to check for efficient membrane trafficking and assess the fluorescence response to ITV. The variant with brightest red and green fluorescence (latter found to have Val and Lys at the fusion site and designated tdFlicR-VK-ASAP) exhibited an increase in red fluorescence and a decrease in green fluorescence when subjected to ITV (10 ms pulses) in Hela cells (Fig. 2B–E).
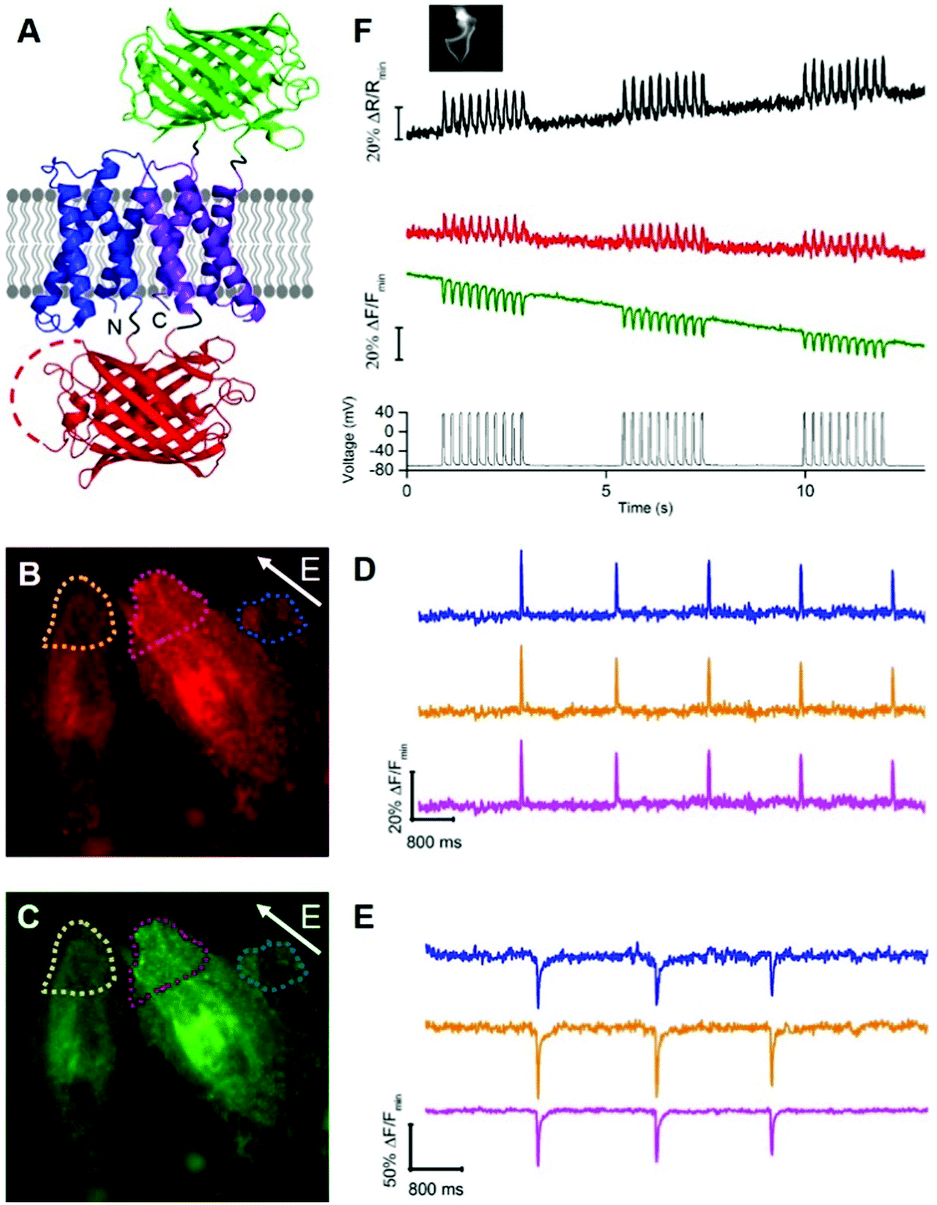 | ||
| Fig. 2 (A) Representation of tdFlicR-ASAP indicator. (B and C) Image of HeLa cells expressing tdFlicR-VK-ASAP in the red channel (B) and green channel (C). (D) tdFlicR-VK-ASAP red fluorescence response of three regions indicated in (B) to ITV (10 ms, 25 V). (E) tdFlicR-VK-ASAP green fluorescence response of the same three regions indicated in (C) to ITV (10 ms, 25 V). (F) Simultaneous patch clamp recording and fluorescence imaging of tdFlicR-VK-ASAP. Gray trace: patch clamp recording (50 ms, 100 mV, 10 Hz). Green trace: raw tdFlicR-VK-ASAP green fluorescence without correction for photobleaching. Red trace: raw tdFlicR-VK-ASAP red fluorescence without correction for photobleaching. Black trace: ratiometric fluorescence change (red/green). Inset: Image of patched HEK 293 cell expressing tdFlicR-VK-ASAP. | ||
Next, we expressed tdFlicR-VK-ASAP under a CMV promoter in HEK 293 cells and used whole cell voltage clamp to modulate membrane potential while recording the fluorescence response in the green and red fluorescence channels simultaneously (see ESI†). In voltage clamped HEK 293 cells, the fluorescence response of tdFlicR-VK-ASAP robustly tracked 10 Hz voltage spikes in both the green and red channels. Green fluorescence decreased by ∼10% ΔF/F0 and red fluorescence increased by ∼5% ΔF/F0, giving a ratiometric change of ∼25% ΔR/R0 (Fig. 2F). Next we expressed tdFlicR-VK-ASAP in dissociated rat hippocampal neurons to test its membrane trafficking in neurons and its ability to respond to spontaneous action potentials. Although tdFlicR-VK-ASAP trafficked to the membrane of neurons, puncta in the cell body were apparent in the red fluorescence channel (Fig. S4A and B, ESI†), as is characteristic of mApple-based indicators. tdFlicR-VK-ASAP successfully reported spontaneous activity in cultured neurons when the green and red channels were imaged separately (Fig. S4C, ESI†). However, we did not observe ratiometric fluorescence responses to spontaneous activity in neurons expressing tdFlicR-VK-ASAP using a widefield fluorescence imaging setup. Using a 200 W lamp as a light source and a beam splitter to separate the two fluorescence channels (see ESI†) resulted in very low fluorescence intensities such that true signals could not be distinguished from the noise (Fig. S4D, ESI†).
The second indicator design strategy was aimed at developing a photoconvertible voltage indicator. Although a plethora of voltage indicators have been reported, none have yet emerged that enable selective highlighting of specific cells such that their changes in membrane potential could be imaged in a second spectral channel. Highlighting could, in principle, be achieved by the use of photoconvertible FPs that irreversibly change their emission wavelength from green to red upon illumination with ∼400 nm light.8 If such highlightable FPs were to be engineered to sense real-time voltage changes, researchers could specifically “highlight” and image the electrical activity of a specific neuron, and then easily re-identify and image the same cell in subsequent experiments within the timeframe of protein turnover.
To engineer a photoconvertible voltage indicator, we fused a cp version of a photoconvertible FP (mMaple)9 to the C-terminus of the S4 α-helix of CiVSD (Fig. 3A). To screen for photoconvertible voltage-sensitive variants, we designed a high-throughput workflow similar to the screen described for tandem dimer voltage indicators (Fig. S5, ESI†). In this case, the E. coli bacterial colony screen was also used to screen for photoconversion efficiency (see ESI†). To identify the optimal composition of the VSD to cpmMaple linker, we randomized the codons for two residues immediately following residue 239 of CiVSD (P240 and P241). Screening of this library for green to red photoconversion, followed by screening for voltage sensitivity, led to the identification of a variant with mutations P240R and P241H. We designated this variant as our first generation fluorescent indicator for voltage imaging – green to red (FlicGR0.1). FlicGR0.1 photoconverted from green to red when illuminated with violet light and exhibited dim green and dim red fluorescence that was responsive to membrane potential changes in mammalian cells. To further improve FlicGR0.1's brightness and voltage sensitivity, we used multiple rounds of directed protein evolution, where each round consisted of construction and screening of a library of thousands of variants (see ESI†). This strategy led to FlicGR1, which was 10 ± 2-fold brighter than FlicGR0.1 (n = 100 colonies) in the green channel and 12 ± 2-fold brighter than FlicGR0.1 (n = 100 colonies) in the red channel following photoconversion (Fig. 3B and C), in the context of bacterial colonies.
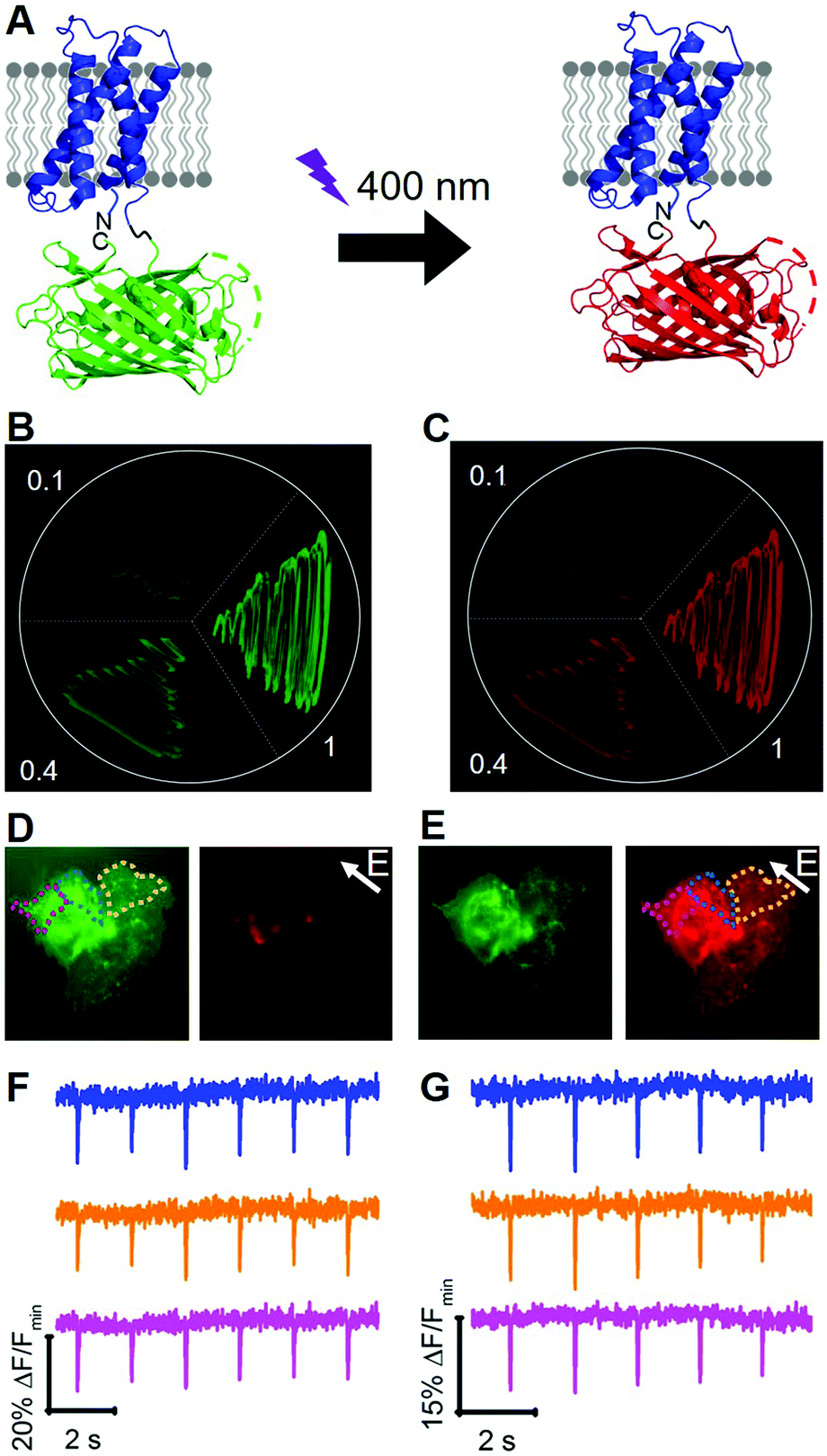 | ||
| Fig. 3 (A) Representation of FlicGR indicator and photoconversion. (B and C) Evolution of FlicGR brightness in E. coli. (B) Green fluorescence image of E. coli expressing FlicGR0.1, 0.4 and 1 before photoconversion. (C) Red fluorescence image of the same E. coli after photoconversion with 405 nm light. (D) Image of HeLa cells expressing FlicGR1 before photoconversion in the green and red channels. Note FlicGR is not fluorescent in the red channel. (E) Image of HeLa cells after photoconversion in the green and red channels. (F) FlicGR1 green fluorescence response of three regions indicated in (D) to ITV (10 ms, 25 V). (G) FlicGR1 red fluorescence response of the same three regions indicated in (E) to ITV (10 ms, 25 V) after photoconversion. | ||
FlicGR1 could be readily photoconverted from green to red when expressed in mammalian cells (Fig. 3D and E) and exhibited a decrease in both the green and red fluorescence channels in response to membrane depolarization (Fig. 3F and G). Compared to the original template, FlicGR1 harbored a total of nine mutations: two mutations in the VSD and seven mutations in cpmMaple (Fig. S6, ESI†). When expressed in dissociated hippocampal neurons, FlicGR1 targeted properly to the plasma membrane of neurons and successfully photoconverted from green to red when illuminated with violet light (Fig. S7 and S8, ESI†). However, FlicGR1 did not produce measurable fluorescence responses to either spontaneous or stimulated activity in neurons. We suspect that FlicGR1 fluorescence response to voltage was insufficient to provide a detectable signal-to-noise ratio to report rapid neuron firing of ∼1 ms action potentials in single-trial recordings.
In summary, we have described our efforts to explore new designs for engineering FP-based voltage indicators. These efforts led to the development of three indicators that serve to validate the potential utility of these design strategies. However, further development will be required to make tools that offer performance on par with previously reported indicators such as Arclight,4e ASAP1,5 and FlicR1.4h
This work was supported by grants from the CFI/Alberta Advanced Education & Technology (K. B.), Alberta Innovates and AIHS (Scientist award to K. B.), CIHR (R. E. C.), NSERC (R. E. C. and K. B.), Brain Canada (R. E. C. and K. B.), and NIH (U01 NS090565-02 to R. E. C.). A. S. A. was supported by a Vanier Canada Graduate Scholarship and an AIHS Studentship.
Notes and references
- (a) S. Chemla and F. Chavane, J. Physiol., 2010, 104, 40–50 CrossRef CAS PubMed; (b) H. Mutoh, W. Akemann and T. Knöpfel, ACS Chem. Neurosci., 2012, 3, 585–592 CrossRef CAS PubMed; (c) F. St-Pierre, M. Chavarha and M. Z. Lin, Curr. Opin. Chem. Biol., 2015, 27, 31–38 CrossRef CAS PubMed.
- (a) K. Ataka and V. A. Pieribone, Biophys. J., 2002, 82, 509–516 CrossRef CAS PubMed; (b) G. Guerrero, M. S. Siegel, B. Roska, E. Loots and E. Y. Isacoff, Biophys. J., 2002, 83, 3607–3618 CrossRef CAS PubMed; (c) T. Knopfel, K. Tomita, R. Shimazaki and R. Sakai, Methods, 2003, 30, 42–48 CrossRef CAS PubMed; (d) R. Sakai, V. Repunte-Canonigo, C. D. Raj and T. Knöpfel, Eur. J. Neurosci., 2001, 13, 2314–2318 CrossRef CAS PubMed; (e) M. S. Siegel and E. Y. Isacoff, Neuron, 1997, 19, 735–741 CrossRef CAS PubMed.
- Y. Murata, H. Iwasaki, M. Sasaki, K. Inaba and Y. Okamura, Nature, 2005, 435, 1239–1243 CrossRef CAS PubMed.
- (a) W. Akemann, H. Mutoh, A. Perron, Y. K. Park, Y. Iwamoto and T. Knöpfel, J. Neurophysiol., 2012, 108, 2323–2337 CrossRef CAS PubMed; (b) B. J. Baker, H. Mutoh, D. Dimitrov, W. Akemann, A. Perron, Y. Iwamoto, L. Jin, L. B. Cohen, E. Y. Isacoff, V. A. Pieribone, T. Hughes and T. Knopfel, Brain Cell Biol., 2008, 36, 53–67 CrossRef CAS PubMed; (c) L. Barnett, J. Platisa, M. Popovic, V. A. Pieribone and T. Hughes, PLoS One, 2012, 7, e43454 CAS; (d) S. G. Gautam, A. Perron, H. Mutoh and T. Knöpfel, Front. Neuroeng., 2009, 2, 14 CrossRef CAS PubMed; (e) L. Jin, Z. Han, J. Platisa, J. R. A. Wooltorton, L. B. Cohen and V. A. Pieribone, Neuron, 2012, 75, 779–785 CrossRef CAS PubMed; (f) A. J. Lam, F. St-Pierre, Y. Gong, J. D. Marshall, P. J. Cranfill, M. A. Baird, M. R. McKeown, J. Wiedenmann, M. W. Davidson, M. J. Schnitzer, R. Y. Tsien and M. Z. Lin, Nat. Methods, 2012, 9, 1005–1012 CrossRef CAS PubMed; (g) A. Lundby, H. Mutoh, D. Dimitrov, W. Akemann and T. Knöpfel, PLoS One, 2008, 3, e2514 Search PubMed; (h) A. S. Abdelfattah, S. L. Farhi, Y. Zhao, D. Brinks, P. Zou, A. Ruangkittisakul, J. Platisa, V. A. Pieribone, K. Ballanyi, A. E. Cohen and R. E. Campbell, J. Neurosci., 2016, 36, 2458–2472 CrossRef CAS PubMed.
- F. St-Pierre, J. D. Marshall, Y. Yang, Y. Gong, M. J. Schnitzer and M. Z. Lin, Nat. Neurosci., 2014, 17, 884–889 CrossRef CAS PubMed.
- Q. Li, S. Wanderling, M. Paduch, D. Medovoy, A. Singharoy, R. McGreevy, C. A. Villalba-Galea, R. E. Hulse, B. Roux, K. Schulten, A. Kossiakoff and E. Perozo, Nat. Struct. Mol. Biol., 2014, 21, 244–252 CAS.
- J. Wu, L. Liu, T. Matsuda, Y. Zhao, A. Rebane, M. Drobizhev, Y.-F. Chang, S. Araki, Y. Arai, K. March, T. E. Hughes, K. Sagou, T. Miyata, T. Nagai, W.-H. Li and R. E. Campbell, ACS Chem. Neurosci., 2013, 4, 963–972 CrossRef CAS PubMed.
- (a) R. Ando, H. Hama, M. Yamamoto-Hino, H. Mizuno and A. Miyawaki, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12651–12656 CrossRef CAS PubMed; (b) H. Mizuno, T. K. Mal, K. I. Tong, R. Ando, T. Furuta, M. Ikura and A. Miyawaki, Mol. Cell, 2003, 12, 1051–1058 CrossRef CAS PubMed.
- (a) H. Hoi, T. Matsuda, T. Nagai and R. E. Campbell, J. Am. Chem. Soc., 2013, 135, 46–49 CrossRef CAS PubMed; (b) A. L. McEvoy, H. Hoi, M. Bates, E. Platonova, P. J. Cranfill, M. A. Baird, M. W. Davidson, H. Ewers, J. Liphardt and R. E. Campbell, PLoS One, 2012, 7, e51314 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supplementary figures. See DOI: 10.1039/c6cc06810c |
| This journal is © The Royal Society of Chemistry 2016 |