
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreation of new guest accessible space under gas pressure in a flexible MOF: multidimensional insight through combination of in situ techniques†
Prashant M.
Bhatt
,
Eustina
Batisai
,
Vincent J.
Smith
and
Leonard J.
Barbour
*
Department of Chemistry and Polymer Science, University of Stellenbosch, Matieland, 7600, South Africa. E-mail: ljb@sun.ac.za; Fax: (+27) 21-808-3360
First published on 25th August 2016
Abstract
Metal–organic frameworks (MOFs) are an important class of porous materials with numerous potential applications. Molecular level understanding of various processes involving MOFs is very important in order to design porous materials with improved properties. Here we describe the elucidation by means of single-crystal X-ray diffraction (SCD) of three different phases of Zn2(bdc)2(bpy) at different pressures of CO2. Moreover, this is a rare example where new space is created in the structure under gas pressure – space that was not previously occupied by guest molecules in any of the related structures. In addition to in situ SCD and sorption analysis, pressure-gradient differential scanning calorimetry (PG-DSC) provides very useful information about the system. This study represents the first instance where PG-DSC has been used to study the sorption behavior of a flexible porous framework.
Phase transformations in solids can be induced by many external stimuli such as heat, light, pressure, etc. In particular, transformations induced by gas pressure have generated much interest: not only do such transformations help us to understand the nature of gas–solid interactions but they can also result in the development of cost-effective industrial processes.1a Solid–gas interactions generally play an important role in such transformations and the nature of the gas is critical for the transformation to proceed. As one of the early examples, Atwood and co-workers reported polymorphic phase transitions from nonporous to porous phases of organic compounds under gas pressure.1b,c Metal–organic frameworks (MOFs),2,3 one of the most investigated classes of material, are also known to undergo such ‘closed’ to ‘open’ structural transformations under gas pressure. The phenomenon is also referred to as ‘gate opening’ or ‘breathing’ and this subclass of MOFs is known as ‘flexible’ or ‘soft’ MOFs. There has been much interest in the ability of flexible MOFs to undergo reversible structural changes upon guest removal, addition or exchange3 as these features can be used for many potential applications. Recently, Long and co-workers have made use of the flexible nature of a MOF to obtain a better working capacity for methane storage than is exhibited by known rigid frameworks.3e
Chen et al. have reported the preparation of the MOF [Zn2(bdc)2(bpy)]·2DMF·H2O (1DMF, also referred to as MOF-508a) from the reaction of 1,4-benzenedicarboxylic acid (bdc), 4,4′-bipyridine (bpy) and Zn(NO3)2·6H2O in DMF/ethanol under solvothermal conditions.41DMF is a twofold interpenetrated 3D MOF with a 2D network of DMF-filled channels. Each framework is based on the zinc paddle-wheel dinuclear secondary building unit (SBU), which propagates in two dimensions via the bdc ligands and in the third dimension by means of bpy pillars. When heated to 120 °C under dynamic vacuum for 24 hours, 1DMF undergoes a single-crystal to single-crystal transformation to the desolvated form [Zn2(bdc)2(bpy)] (2, previously referred to as MOF-508b).4 Although the overall framework connectivity and degree of interpenetration do not change during this transition, the structure of 2 (Fig. 1a) is very different from that of 1DMF. Considerable distortion of the zinc paddle-wheel SBU occurs, with bending in the coordination geometry of the bpy linkers in 2. This results in compression of the framework, with a 16.7% reduction in the volume of the crystal. A map of the guest-accessible volume in 2 (Fig. 1b) reveals the presence of discrete 66 Å3 voids per host formula unit with no permanent channels between them (see ESI† for a comment on void volume calculations). The conversion from 1DMF to 2 is known to be reversible when 2 is soaked in a DMF/ethanol solution for one day.4 In the original report describing 1DMF and 2 the authors presumed that 2 undergoes a single phase transition, converting to the gas-included analogue of 1DMF during CO2 sorption. This assumption was based on their sorption isotherm and other information available at that time.4
 | ||
| Fig. 1 Perspective views of the twofold interpenetrated networks (colored blue and red) of 2, 1CO2 and 3CO2. (a) 2 viewed along the bpy ligand N⋯N axis and (b) 0D voids in 2 viewed perpendicular to the bpy ligands. (c) 1CO2 viewed along the bpy ligands and (d) guest-accessible 2D channels in 1CO2. (e) 3CO2 viewed along the bpy ligands and (f) the guest-accessible space corresponding to that of 1CO2. The 2D channel networks in 3CO2 are connected to each other by newly formed guest-accessible channels (not shown but the included CO2 molecules within the new space are shown in green). Note the difference in the coordination geometry of the ligands and the evolution of guest-accessible space concomitant with the structural transformations from 0D in 2 to 2D in 1CO2 to 3D in 3CO2. Guest-accessible space was mapped using a probe radius of 1.52 Å. | ||
For more insight, we carried out CO2 sorption experiments on 2 at 25 °C in the pressure range 0–25 bar, as shown in Fig. 2 (top). The amount of gas adsorbed at room temperature and high pressure is comparable to that reported in the literature for low temperature and low pressure.4 However, the sorption isotherm at room temperature is much better resolved compared to that recorded at 195 K and shows two clear inflection points, suggesting that the system undergoes at least two phase transitions under CO2 pressure. It is evident from Fig. 2, that a comparatively small amount of sorption occurs (≈0.8 moles of CO2 per formula unit) up to 7 bar of CO2 pressure. This sorption trend is consistent with the existence of 66 Å3 voids per host forumla unit in 2; each void could conceivably accommodate a maximum of one molecule of CO2. However, after 7 bar the host structure ‘opens up’ and tends toward taking up an additional two molecules of CO2 per host formula unit. Around 10 bar a second, smaller inflection in the sorption isotherm suggests another structural change in the system to accommodate a further molecule of CO2 per host formula unit. After this point the system slowly reaches saturation in the range of 10 to 25 bar (i.e. 3.82 molecules of CO2 per host formula unit). The desorption cycle also features two inflection points, as well as moderate hysteresis; it is reasonable to assume that the two CO2 pressure-induced ‘gate opening’ phase transformations are reversed during desorption. The onset pressures for the reverse ‘gate closing’ phase transitions are approximately 8 and 6 bar – owing to hysteresis, these are somewhat lower values than recorded for the corresponding transitions due to CO2 uptake.
 | ||
| Fig. 2 CO2 sorption isotherm for [Zn2(bdc)2(bpy)] recorded at 25 °C (top); inflection points in the isotherm suggest phase transitions (i.e. gate-opening events). PG-DSC of [Zn2(bdc)2(bpy)] at 25 °C using a constant pressure gradient of 0.2 bar min−1 in the pressure range 2 to 23 bar of CO2 (bottom). | ||
In order to obtain structural insight the pressure-induced phases were investigated crystallographically while subjecting the crystals to controlled CO2 pressures in situ.5 Single crystals of 1DMF were prepared according to the literature procedure4 and activated to yield the marginally porous structure 2 (described above). A crystal suitable for SCD analysis was selected and its structures were determined under various pressures of CO2 using an environmental gas cell developed by us.5a The marginally porous structure 2 with its narrow 0D voids (Fig. 1b) remains unchanged in the pressure range 0 to 8 bar of CO2 at room temperature (25 °C). When the CO2 pressure is raised to approximately 10 bar, 2 (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) undergoes a single-crystal to single-crystal transformation to yield 1CO2 (also triclinic, P).
) undergoes a single-crystal to single-crystal transformation to yield 1CO2 (also triclinic, P).
The structure of 1CO2 is similar to that of 1DMF but with CO2 as the guest instead of DMF. During the transformation from 2 to 1CO2 the grid formed by the dinuclear zinc paddle-wheel SBUs and the bdc linkers distorts only slightly, with the grid angle changing from 72.74(3)° to 75.57(2)°. However, the most significant structural difference involves the coordination geometry of the bpy ligand, which changes from bent to almost linear. This affects the relative positions of the adjoining grids, which in turn results in changes in the relative positions of the two interpenetrating networks and also the transformation from a ‘closed’ to an ‘open’ structure (Fig. 1a and c, respectively). The transformation is thus accompanied by conversion of the narrow 0D voids of 2 to the comparatively larger guest-accessible channels of 1CO2, which are interconnected in two dimensions (Fig. 1b and d, respectively). Since these 2D channels (Channels A) are not linked along the third dimension the structure can be described as possessing segregated 2D channels (Fig. S6b, ESI†).
Similar to 1DMF, a considerable amount of electron density is apparent in the channels of 1CO2 but the density could not be modelled as individual CO2 molecules. When the CO2 pressure is increased to 20 bar, 1CO2 (triclinic, P) converts to a new form 3CO2 (monoclinic, P21/c) while retaining its crystal singularity. Form 3CO2 can also be obtained by cooling the crystal of 1CO2 to −20 °C (the initial CO2 pressure at room temperature was 10 bar but, owing to the small size of the environmental gas cell, the final pressure at −20 °C could not be measured). Although the differences in the conformations of the connecting ligands in the structures of 1CO2 and 3CO2 are relatively subtle, these differences have a marked impact on the overall structure and porosity of the material. The largest structural differences between 1CO2 and 3CO2 arise from a change in the shape of the 2D grid formed by the dinuclear zinc paddle-wheel SBUs and the bdc linkers. In the structure of 1CO2, the grid units resemble a rhombus while in the structure of 3CO2 the linkers are comparatively straight, resembling a square (Fig. 1c and e, respectively). Another difference between the structures of 1CO2 and 3CO2 is apparent in the torsion angles of the carboxylate groups of the bdc linkers (see Table S2, ESI†). An even more profound change is observed in the bdc linkers along the crystallographic a axis of 3CO2, where the torsion angles change from 8.4(8)° and 3.8(6)° to 9.5(3)° and 8.0(3)°. There is also a moderate change in the dihedral angle between the two pyridyl rings in the bpy linker when the structure changes from 1CO2 to 3CO2, as shown in Fig. 1d, f and Fig. S7 and Animation S1 (ESI†). Although the individual changes in the abovementioned angles are not very large, in combination with the changes in the grid angles and the other subtle structural changes they ultimately result in the creation of new guest-accessible space in 3CO2 (Channel B, Fig. S6C, ESI†), which accommodates two extra CO2 molecules for each unique new channel, or an additional CO2 molecule per formula unit (Fig. 1d, f and Fig. S7 and Animation S1, ESI†). It is worth noting that the region corresponding to that occupied by CO2 in the newly formed channels (Channel B, Fig. S6C, ESI†) of 3CO2 is unoccupied in 1CO2; i.e. no residual electron density is observed in this space. Moreover, the space is not large enough to accommodate any guest molecules in 1CO2. As shown in Fig. S6c (ESI†), a map of the solvent-accessible surface of 3CO2 shows that the newly formed channels B interconnect the 2D channels (Channel A) that are also present in 1CO2.
Although the guest molecules in the larger channels of 3CO2 (at −20 °C and 10 bar initial pressure) only yield diffuse electron density distribution patterns in the crystallographic analysis (as was the case for both 1DMF and 1CO2), it was possible to model the CO2 molecules in the smaller channels of 3CO2 (Channels B). It therefore appears that the confined environment of the smaller channels facilitates immobilization and crystallographic modeling of CO2. Site occupancy refinement suggests an occupancy of approximately 80% for the CO2 molecules in the smaller channels. The geometry of the unique CO2 molecule (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths of 1.09(2) and 1.18(2) Å and a bond angle of 173(2)°) is not ideal but corresponds well with that of structures containing CO2 in the Cambridge Structural Database (CSD).5c,6,7 Each interconnecting channel contains two symmetry-related CO2 molecules arranged to form an offset dimer that maximizes the interaction between the electron rich oxygen atom (δ−) and the electron deficient carbon atom (δ+), as shown in Fig. 3. The intermolecular C⋯O distance between the two CO2 molecules is 3.13(2) Å; similar CO2 dimers are present in the crystal structure of solid CO2,8 as well as in some of the other examples present in the CSD.6 The central carbon atom of each CO2 molecule also forms a relatively short C⋯O contact of 3.07(2) Å with one of the carboxylate oxygen atoms of a bdc linker, as shown in Fig. 3.
O bond lengths of 1.09(2) and 1.18(2) Å and a bond angle of 173(2)°) is not ideal but corresponds well with that of structures containing CO2 in the Cambridge Structural Database (CSD).5c,6,7 Each interconnecting channel contains two symmetry-related CO2 molecules arranged to form an offset dimer that maximizes the interaction between the electron rich oxygen atom (δ−) and the electron deficient carbon atom (δ+), as shown in Fig. 3. The intermolecular C⋯O distance between the two CO2 molecules is 3.13(2) Å; similar CO2 dimers are present in the crystal structure of solid CO2,8 as well as in some of the other examples present in the CSD.6 The central carbon atom of each CO2 molecule also forms a relatively short C⋯O contact of 3.07(2) Å with one of the carboxylate oxygen atoms of a bdc linker, as shown in Fig. 3.
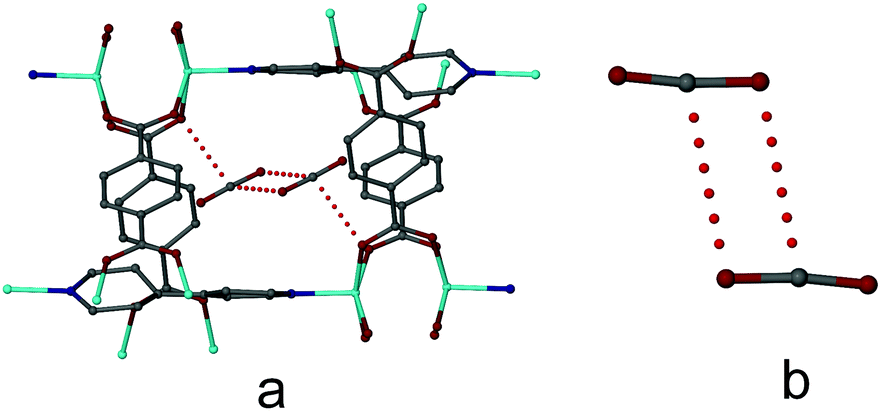 | ||
| Fig. 3 (a) C⋯O interaction between the CO2 carbon atoms and a carboxylate oxygen atom of the host network in 3CO2; (b) C⋯O interaction in the CO2 dimer. | ||
There are only a few reports in the literature of nonporous structures that transform to porous phases under gas pressure.1b,c,3 These processes involve dynamic changes in the host structure in order to accommodate guest molecules, and they seldom occur as single-crystal to single-crystal transformations. Furthermore, in most of these cases a solvate equivalent of the open-framework gas-loaded structure is known, and the nonporous “collapsed” phase is generally prepared by removing the solvent. The collapse of the host framework upon solvent removal is therefore reversible (provided that the gas pressure is sufficient to effect the transformation) and the gas molecules ultimately occupy the space previously templated by the solvent molecules. Indeed, we have observed that this phenomenon occurs for the transformation of solvate 1DMF into nonporous 2, followed by the transformation of 2 into gas-loaded 1CO2. Application of additional CO2 gas pressure results in transformation to 3CO2, with the creation of new space that had not previously been occupied by guest molecules in any of the known solvated structures involving this framework. To our knowledge, the only other slightly similar example in the literature is the system [M2(O2CPh)4(pyrazine)]n (M = Rh2+ or Cu2+), widely studied by Takamizawa and others.6,9
We considered the desirability of monitoring gas pressure-induced phase transformations or flexibility in MOFs and other porous materials in a time efficient manner using a scanning technique akin to differential scanning calorimetry (DSC), which is commonly employed to observe thermally-induced phase transitions (melting, crystallization, polymorphic conversion, chemical reaction, etc.). Such capability could prove very useful for fast and efficient screening for flexibility and phase transitions of MOFs and other solids in response to gas pressure. Although DSC has already been used to determine the sorption enthalpy for porous materials in a few cases,10 in all of these examples one or more measurements were made by instantaneous dosing rather than using a scanning approach. Indeed, the limitations of DSC in non-scanning mode for such experiments have been acknowledged in the literature.11 DSC has also been used in scanning mode to investigate phase transformations of solids under gas pressure either by ramping the temperature at constant gas pressure6 or by flowing gas at a constant rate at cryogenic temperatures.11 However isothermal DSC analysis while increasing the pressure gradually and at a constant rate has not been realized to date. We therefore developed a method involving pressure-gradient DSC (PG-DSC) and validated the technique using the present MOF. A prototype PG-DSC system was constructed by coupling commercially available modules, including a gas syringe pump (Teledyne ISCO 260D) and a DSC analyser (Setaram μDSC7 Evo). In order to increase the gas pressure at a desired constant rate the syringe pump is controlled by a computer running software that was developed by us. We used PG-DSC to study the present system [Zn2(bdc)2(bpy)] since this material is now well characterized structurally by in situ crystallography. Fig. 2 (bottom) shows the PG-DSC trace in the range 2–23 bar; two exotherms are observed in the range of 7–10 bar (large) and 11–15 bar (small, broad) during sorption and two endotherms are evident at the shifted pressure range of 7–5 bar (small) and 5–3 bar (large) during desorption. The data are consistent with our results obtained from sorption analysis and in situ SCD.
The PG-DSC peaks are mostly due to the heats of sorption and desorption, combined with a comparatively very small component from the host structural transformations. Therefore the PG-DSC experiments provide valuable information about gate opening/closing onset pressures and gas uptake/release hysteresis in a time-efficient manner. Moreover, it is possible to determine the shifts in these pressures with temperature; the PG-DSC traces for 2 at four different temperatures (0, 15, 25 and 45 °C) are shown in Fig. S12 (ESI†). It is evident that the gate opening pressure and extent of hysteresis increase with temperature. Furthermore, gate opening pressures for a system exposed to different gases can be compared in a rapid and efficient way using PG-DSC. To demonstrate this, PG-DSC experiments were carried out for 2 using CH4 as the test gas and gate-opening occurred in the broad ranges of 30–50 and 60–90 bar at 0 and 25 °C, respectively (See ESI,† Fig. S13 and S14). After validating the PG-DSC technique using the present system we also used the method to detect gas-induced phase transformations for other reported flexible systems (see ESI†).
In summary, the present work describes the observation of three different structures by SCD at different pressures of CO2 in the system Zn2(bdc)2(bpy). Moreover, this is a rare example where new guest accessible space is formed under gas pressure: space that was not previously occupied by guest molecules in any of the related structures. The newly-developed technique of PG-DSC was successfully validated using the present system and then also applied to other systems. PG-DSC offers fast and efficient screening of materials for gas-induced phase transformations and efforts to determine useful thermodynamic data from this technique are currently in progress.
The authors thank the National Research Foundation of South Africa for financial support. PB also thanks the Claude Leon Foundation for a fellowship.
Notes and references
-
(a) J. Tian, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 1399–1404 CrossRef CAS PubMed
; (b) P. K. Thallapally, B. Peter McGrail, S. J. Dalgarno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146–150 CrossRef CAS PubMed
-
(a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
-
(a) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS PubMed
-
(a) T. Jacobs, G. O. Lloyd, J.-A. Gertenbach, K. K. Müller-Nedebock, C. Esterhuysen and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 124, 4997–5000 CrossRef
- S. Takamizawa, E. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 115, 4467–4470 CrossRef
-
(a) T. K. Maji, G. Mostafa, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2005, 127, 17152–17153 CrossRef CAS PubMed
- C. S. Yoo, H. Kohlmann, H. Cynn, M. F. Nicol, V. Iota and T. LeBihan, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104103 CrossRef
- S. Takamizawa, E. Nataka, T. Akatsuka, R. Miyake, Y. Kakizaki, H. Takeuchi, G. Maruta and S. Takeda, J. Am. Chem. Soc., 2010, 132, 3783–3792 CrossRef CAS PubMed
-
(a) S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519–13521 CrossRef CAS PubMed
- P. L. Llewellyn and G. Maurin, C. R. Chim., 2005, 8, 283–302 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: SCD, sorption, PG-DSC, PXRD, CCDC 1429431–1429435. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06709c |
| This journal is © The Royal Society of Chemistry 2016 |