
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable bromoallene oxides†
D.
Christopher Braddock
 *,
Areeb
Mahtey
,
Henry S.
Rzepa
and
Andrew J. P.
White
*,
Areeb
Mahtey
,
Henry S.
Rzepa
and
Andrew J. P.
White
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.braddock@imperial.ac.uk; Fax: +44 (0)2075945805; Tel: +44 (0)2075945772
First published on 30th August 2016
Abstract
The first stable bromoallene oxides were obtained by the DMDO epoxidation of 1-bromo-1,3-di-tert-alkylallenes, producing the first crystalline allene oxide of any kind. The epoxidations are regioselective for the bromine-bearing Δ1,2 alkene, and also face selective producing single diastereomer E-olefin products.
The allene oxide functional group and its valence tautomeric cyclopropanone and oxyallyl cation forms are of significant interest.1,2 The first room-temperature stable allene oxide was reported in 1968 by peracid epoxidation of 1,3-di-tert-butylallene 1 to give epoxide 2.‡
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 It was subsequently converted into its corresponding isomeric cyclopropanone 3 on heating thereby establishing their relative thermodynamic stabilities. However, despite this early success, only a handful of further allene oxides have been successfully characterised.4–10 Nevertheless, these studies have helped to establish that stable allene oxides can be obtained when (i) the epoxide is kinetically stabilised by steric protection against SN2 attack, and (ii) when cyclopropanone formation via the presumed intermediacy of an oxyallyl cation is non-favoured.1c
3 It was subsequently converted into its corresponding isomeric cyclopropanone 3 on heating thereby establishing their relative thermodynamic stabilities. However, despite this early success, only a handful of further allene oxides have been successfully characterised.4–10 Nevertheless, these studies have helped to establish that stable allene oxides can be obtained when (i) the epoxide is kinetically stabilised by steric protection against SN2 attack, and (ii) when cyclopropanone formation via the presumed intermediacy of an oxyallyl cation is non-favoured.1cWe have recently reported11 that DMDO epoxidation of 3-n-alkyl-1-bromo-allenes 4 give rise directly to α,β-unsaturated carboxylic acids 5 by presumed spontaneous rearrangement of bromo-allene oxides – an unprecedented functional group in the literature12 – to bromocyclopropanones and subsequent Favorskii rearrangement. In that work, an analysis of the mechanism revealed that epoxidation of either olefin of the bromoallene would lead to the same product and so the site of initial epoxidation could not be identified. With the intention of identifying the preferred position of epoxidation in bromoallenes, we now report the first stable bromoallene oxides.

With the above literature precedents in mind, we selected known 1-bromo-1,3-di-tert-butylallene 6 – reported as a colourless liquid – as a suitable substrate for epoxidation.13 However, in our hands it proved to be inseparable from its propargylic bromide isomer, was found to be somewhat volatile, and consequently epoxidation experiments proved inconclusive. Instead, we targeted novel adamantyl-containing bromoallenes 7–9, which proved to overcome these difficulties (vide infra).

Bromoallene 7 was prepared by the initial lithiation of terminal alkyne 10 and addition to adamantanecarboxaldehyde§ to give propargylic alcohol 11 (Scheme 1). Temperature control in this transformation proved critical, otherwise significant quantities of ketone 12 and 1-adamantanemethanol were formed.¶|| Using the conditions of Corey,13 alcohol 11 was transformed into bromoallene 7 along with the expected, but now separable, propargylic bromide 13.** Next, the action of the carbon tetrachloride–triphenylphosphine reagent combination14 on adamantanecarboxaldehyde as a α,α,α-trisubstituted aldehyde provided ready access to vinyldichloride 1415 free from any complicating dichloroalkane formation.16 Corey–Fuchs type acetylide anion formation,17 and in situ trapping18 with pivaldehyde and adamantanecarboxaldehyde gave propargylic alcohols 15 and 17 respectively in good yields.†† Bromoallenes 8 and 9 were subsequently obtained by subsequent reaction with thionyl bromide along with the corresponding expected, and again separable, propargylic bromides 16 and 18.‡‡
 | ||
| Scheme 1 Synthesis of bromoallenes 7–9 (Ad = 1-adamantyl). | ||
The incorporation of adamantyl groupings rendered the three bromoallenes 7–9 to be solid, bromoallene 9 proved to be crystalline, and its structure was solved by X-ray crystallography.‡§§ With these compounds in hand, attention turned to epoxidation.¶¶
In the event, epoxidation of each of bromoallenes 7–9 with DMDO19 provided monoepoxidation products as E-1-bromoallene oxides 19–21|||| as single diastereoisomers respectively, along with bromodiketones 22–24,*** and recovered bromoallene starting materials after column chromatography (Scheme 2).††† Importantly, under the conditions of the experiment and the isolation and purification regime, there was no evidence for the formation of any corresponding α,β-unsaturated carboxylic acids via intersecting bromoallene oxide–bromocyclopropanone–Favorskii rearrangement.11 These experiments therefore establish that (i) bromoallene oxides of this type are kinetically stabilised with respect to rearrangement by the bulky alkyl substituents; (ii) for these bromoallenes, epoxidation occurs preferentially at the bromine-bearing Δ1,2 alkene and (iii) epoxidation occurs with complete face selectivity away from the alkyl grouping at C-3 of the allene.‡‡‡ Moreover, the uniformity of the product distributions obtained with the different bromoallenes 7–9 show that the tert-butyl and adamantyl groups behave as effectively identical and interchangeable substituents in this situation, but with the advantage that the incorporation of an adamantyl group increases the prospect of crystallinity. Indeed, bromoallene oxide 19 proved to be crystalline and its structure was determined by X-ray crystallography (Fig. 1). To the best of our knowledge this is the first X-ray crystallographic structural elucidation of any allene oxide.§§§20
 | ||
| Scheme 2 Epoxidation of bromoallenes 7–9. | ||
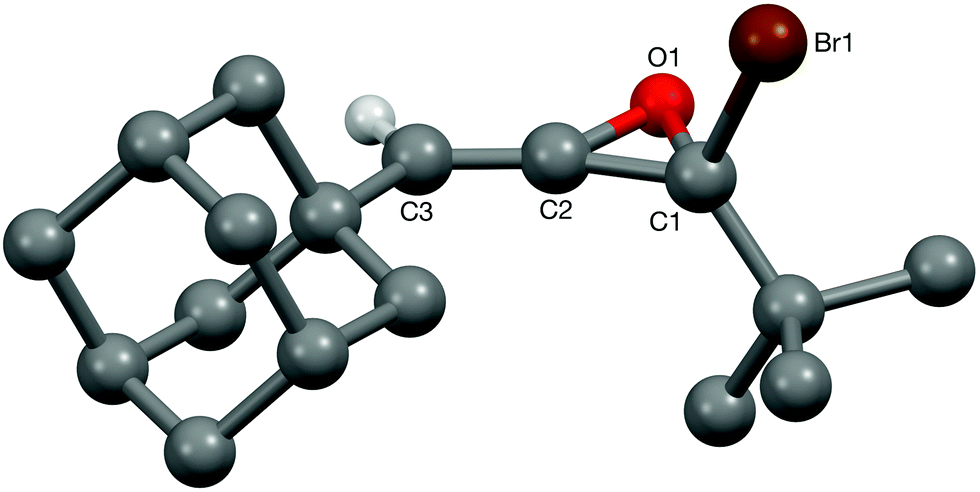 | ||
| Fig. 1 X-ray crystal structure of bromoallene oxide 19. | ||
We probed the origins of the observed regioselectivity and diastereoselectivity using DFT theory (ωB97XD/TZVP,QZVP/C PCM = acetone as continuum solvent).21 An NBO analysis on representative reactant bromoallene 6 reveals the Δ2,3 localized occupied π-orbital is higher in energy (by 0.014 Hartree) than the Δ1,2 orbital, suggesting that the intrinsic electronic nucleophilic reactivity of the allene is at the 2,3 position. However, transition state models of the reaction with DMDO as electrophile reveals the 1,2 isomers are 1.2 (QZVP) or 1.4 (TZVP) kcal mol−1 lower in  for bromoallene 6 and 1.2 (TZVP) for bromoallene 9. For the simplest possible bromoallene (H2C
for bromoallene 6 and 1.2 (TZVP) for bromoallene 9. For the simplest possible bromoallene (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCHBr, not shown) the 2,3 transition state at the π-face anti-periplanar to the Br is favoured by 0.4 kcal mol−1 over the 1,2 isomer and by 1.3 kcal mol−1 over 2,3-syn-periplanar attack (TZVP). Taken together, these calculations reveal that although the bromine atom is inherently deactivating for the Δ1,2 olefin, it also acts to sterically protect the syn-periplanar face of the Δ2,3 olefin. For bromoallenes 7–9, since the bulky alkyl group on C-1 protects the other face of Δ2,3 olefin and the C-3 alkyl group inhibits Z-1-bromoallene oxide formation, Δ1,2 attack leading to the E-1-bromoallene oxides 19–21 emerges as the most facile.
CCHBr, not shown) the 2,3 transition state at the π-face anti-periplanar to the Br is favoured by 0.4 kcal mol−1 over the 1,2 isomer and by 1.3 kcal mol−1 over 2,3-syn-periplanar attack (TZVP). Taken together, these calculations reveal that although the bromine atom is inherently deactivating for the Δ1,2 olefin, it also acts to sterically protect the syn-periplanar face of the Δ2,3 olefin. For bromoallenes 7–9, since the bulky alkyl group on C-1 protects the other face of Δ2,3 olefin and the C-3 alkyl group inhibits Z-1-bromoallene oxide formation, Δ1,2 attack leading to the E-1-bromoallene oxides 19–21 emerges as the most facile.
In conclusion, we have demonstrated that stable bromoallene oxides can be obtained by DMDO epoxidation of 1-bromo-1,3-di-tert-alkylallenes, and the first X-ray crystal structural elucidation of any allene oxide has thereby been accomplished. The epoxidation for these bromoallenes is selective for the bromine-bearing Δ1,2 alkene, and with exclusive face selectivity to produce E-1-bromoallene oxides.¶¶¶ The thermal stability of these bromoallene oxides is the subject of current investigations and will be reported in due course.
We thank Mr Jaren Soo for preliminary investigations with compound 6.
Notes and references
- For reviews see: (a) G. L'abbé, Angew. Chem., Int. Ed. Engl., 1980, 19, 276–289 CrossRef; (b) W. Smadja, Chem. Rev., 1983, 83, 263–320 CrossRef CAS; (c) T. H. Chan and B. S. Ong, Tetrahedron, 1980, 36, 2269–2289 CrossRef CAS.
- For computational studies see: B. A. Hess, Jr., U. Eckart and J. Fabian, J. Am. Chem. Soc., 1998, 120, 12310–12315 CrossRef and references cited therein.
- (a) R. L. Camp and F. D. Greene, J. Am. Chem. Soc., 1968, 90, 7349 CrossRef CAS . A contemporaneous report on the peracid epoxidation of isomeric 1,1-di-tert-butylallene showed that 2,2-di-tert-butylcyclopropanone was formed instead: ; (b) J. K. Crandall and W. H. Machleder, J. Am. Chem. Soc., 1968, 90, 7347–7349 CrossRef CAS . 1,1-Di-tert-butylallene oxide was later characterised in solution after low temperature epoxidation (ref. 4).
- 1,1,3-Tri-tert-butylallene oxide reported as a 10:1 mixture with an oxetane by glpc, and 1,3-di-tert-butyl-1-methylallene oxide as the major component of a four component mixture: J. K. Crandall, W. W. Conover, J. B. Komin and W. H. Machleder, J. Org. Chem., 1974, 39, 1723–1729 CrossRef CAS . These trisubstituted allene oxides were reported to be resistant to conversion to their isomeric cyclopropanones.
- 1-tert-Butylallene oxide as a ‘reasonably pure compound’ by reduced pressure bulb-to-bulb distillation: (a) T. H. Chan and B. S. Ong, J. Org. Chem., 1978, 53, 2994–3001 CrossRef and references cited therein. This allene oxide was reported to polymerise on standing in solution. This allene oxide has been prepared as a single enantiomer: ; (b) T. Konoike, T. Hayashi and Y. Araki, Tetrahedron: Asymmetry, 1994, 5, 1559–1566 CrossRef CAS.
- For a stable 3-tert-butylallene oxide from a fulvene endo-peroxide isolated by preparative tlc see: I. Erden, J. Drummond, R. Alstad and F. Xu, Tetrahedron Lett., 1993, 34, 1255–1258 CrossRef CAS.
- Some non tert-butylallene oxides could be generated photochemically and detected by 1H NMR methods in solution: (a) L. E. Breen, N. P. Schepp and C.-H. E. Tan, Can. J. Chem., 2005, 83, 1347–1351 CrossRef CAS . See also: ; (b) M. D. Clay, J. Durber and N. P. Schepp, Org. Lett., 2001, 3, 3883–3886 CrossRef CAS PubMed; (c) M. W. Konecny and N. P. Schepp, Org. Biomol. Chem., 2009, 7, 4437–4443 RSC and references cited therein.
- For the isolation and characterization of a natural allene oxide as an unstable intermediate in the metabolism of lipid hydroperoxides see: (a) A. R. Brash, S. W. Baertschi, C. D. Ingram and T. M. Harris, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3382–3386 CrossRef CAS PubMed and references cited therein. For E vs. Z isomers see: ; (b) A. R. Brash, W. E. Boeglin, D. F. Stec, M. Voehler, C. Schneider and J. K. Cha, J. Biol. Chem., 2013, 288, 20797–20806 CrossRef CAS PubMed . See also: ; (c) N. V. Medvedeva, S. K. Latypov, A. A. Balandina, L. S. Mukhtarova and A. N. Grechkin, Russ. J. Bioorg. Chem., 2005, 31, 595–596 CrossRef CAS.
- For representative use of allene oxides as intended transient intermediates see: (a) M. Shipman, H. R. Thorpe and I. R. Clemens, Tetrahedron, 1998, 54, 14265–14282 CrossRef CAS; (b) I. R. Clemens, M. Shipman and H. R. Thorpe, Synlett, 1995, 1065–1066 CrossRef CAS; (c) S. J. Kim and J. K. Cha, Tetrahedron Lett., 1988, 29, 5613–5616 CrossRef CAS and references cited therein.
- For the first report of a stable spirodiepoxide (from bisepoxidation of allenes) see: (a) J. K. Crandall, W. H. Machleder and M. J. Thomas, J. Am. Chem. Soc., 1968, 90, 7346–7347 CrossRef CAS . For a recent representative use of spirodiepoxides in cascade reactions see: ; (b) R. Sharma, M. Manpadi, Y. Zhang, H. Kim, N. G. Ahkmedov and L. J. Williams, Org. Lett., 2011, 13, 3352–3355 CrossRef CAS PubMed.
- D. C. Braddock, J. Clarke and H. S. Rzepa, Chem. Commun., 2013, 49, 11176–11178 RSC.
- Ethyl 2-bromo-3-(diphenylmethylene) oxirane-2-carboxylate (i.e., a bromoallene oxide ester) was reported in a study of ketenes and aliphatic diazo compounds: H. Staudinger and T. Reber, Helv. Chim. Acta, 1921, 4, 3–23 CrossRef CAS.
- E. J. Corey and N. W. Boaz, Tetrahedron Lett., 1984, 25, 3055–3058 CrossRef CAS.
- R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801–811 CrossRef.
- This useful building block has only been reported once previously (by a different method): J. Buendia, B. Darses and P. Dauban, Angew. Chem., Int. Ed., 2015, 54, 5697–5701 CrossRef CAS PubMed.
- The CCl4–PPh3 combination on aldehydes typically gives rise to dichloromethane and dichloroalkene mixtures: R. Rabinowitz and R. Marcus, J. Am. Chem. Soc., 1962, 84, 1312–1313 CrossRef CAS.
- There are surprisingly only limited literature precedents for the alkyl lithium mediated conversion of 1,1-dichloroalkenes into terminal alkynes. For a representative example see: F. Bertrand, B. Quiclet-Sire and S. Z. Zard, Angew. Chem., Int. Ed., 1999, 38, 1943–1946 CrossRef CAS.
- There is only one example in the literature that traps the presumed lithium acetylide from the action of alkyl lithiums on 1,1-dichloroalkenes with aldehydes as electrophiles: Z. Li and S. Z. Zard, Org. Lett., 2009, 11, 2868–2871 CrossRef CAS PubMed.
- R. W. Murray and M. Singh, Org. Synth., 1997, 74, 91 CrossRef CAS.
- A lithiated oxirane with considerable asymmetry has recently been characterised by crystallography: A. Salomone, F. M. Perna, A. Falcicchio, S. O. Nilsson Lill, A. Moliterni, R. Michel, S. Florio, D. Stalke and V. Capriati, Chem. Sci., 2014, 5, 528–538 RSC.
- For all managed computational research data, including input and output files, see: D. C. Braddock, A. Mahtey, H. S. Rzepa and A. J. P. White, Imperial College HPC data repository, 2016. Collection doi: http://10.14469/HPC/442 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterising data for 7–9, 11–24; copies of 1H and 13C NMR spectra for all compounds including NOE, DEPT-135, correlation spectroscopy and bromide induced isotopic shifts where appropriate. CCDC 1485934 (9) and 1485935 (19). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06395k |
| ‡ Interestingly, a definitive structure for allene oxide 2 was not given in the original report and two geometrical isomeric structures were proffered. Our results (vide infra) support this to be the E-allene oxide as drawn. |
| § Readily prepared from 1-adamantanemethanol by PCC oxidation.‡ |
| ¶ See ESI† for experimental details. See also ref. 21. |
|| We invoke an unprecedented irreversible hydride transfer from the initially formed secondary lithium alkoxide AdCH(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)OLi to still unreacted AdCHO in a concerted six-membered TS to explain this observation. In this process steric compression of the adamantyl-bearing sp3 centre is relieved, and the formation of conjugated ynone 12 acts as a thermodynamic driving force. Formation of the lithium alkoxide from alcohol 11 by the action of n-BuLi, and subsequent addition of AdCHO gave also ynone 12. CtBu)OLi to still unreacted AdCHO in a concerted six-membered TS to explain this observation. In this process steric compression of the adamantyl-bearing sp3 centre is relieved, and the formation of conjugated ynone 12 acts as a thermodynamic driving force. Formation of the lithium alkoxide from alcohol 11 by the action of n-BuLi, and subsequent addition of AdCHO gave also ynone 12. |
| ** Unwanted bromide 13 was found to slowly isomerise to bromoallene 7 on standing as a neat liquid, or this could be induced by the action of LiCuBr2 in refluxing THF. As a representative experiment, after 20 h at reflux, allene 7 and bromide 13 were obtained as a 1.35:1 mixture respectively. |
| †† Temperature control was found to be essential to prevent hydride transfer to give products of the type RCOCCAd and RCH2OH. |
| ‡‡ Bromides 16 and 18 also isomerised on standing (tlc analysis) into their respective bromoallenes. |
| §§ The regioisomeric structures of bromoallenes 7 and 8 follow by the nature of the SN2′ reaction in which they are formed (ref. 13). These structures are further supported by characteristic NOE enhancements and HMBC correlations.‡ |
| ¶¶ At the outset, in a simplistic model, we predicted that epoxidation of a bromoallene would be electronically disfavoured at the Δ2,3 alkene position due to the deactivating donation of its π-cloud into the periplanar C–Br σ* orbital. However, it was unclear whether the mesomeric activating release of a bromine lone pair into the Δ1,2 alkene would overcome its inductive deactivating effect. |
|||| The bromoallene oxides 19–21 showed characteristic allene oxide vinyl resonances in their 1H NMR spectra (CDCl3) at 4.84, 4.97 and 4.83 ppm respectively allowing immediate identification of regiochemistry of epoxidation. Their respective 13C NMR resonances (CDCl3) at C-3 (100.2, 100.3, 100.4 ppm) match perfectly with an allene oxide of the type tBu![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HC–(–O–)CHR (100.3 ppm, ref. 6). Characteristic IR stretches for all bromides 19–21 at 1806 cm−1 show them to be allene oxides (see ref. 1c). HC–(–O–)CHR (100.3 ppm, ref. 6). Characteristic IR stretches for all bromides 19–21 at 1806 cm−1 show them to be allene oxides (see ref. 1c). |
| *** We invoke the formation of bromodiketones 22–24via further epoxidation of bromoallene oxides 19–21 to give their respective bromospirodiepoxides, followed by ring-opening rearrangement to the diketones with bromide anion loss from C-1 and gain at C-3 (for the ring-opening rearrangement of spirodiepoxides of trisubstituted allenes with halide anions see ref. 10b). They were assigned these structures on the basis of their characteristic diketone stretches in their IR spectra (22: 1713, 1694 cm−1; 23: 1719, 1690 cm−1; 24: 1717, 1692 cm−1), the presence of two carbonyl resonances in their 13C NMR spectra (22: 206.3, 190.9 ppm; 23: 206.3, 191.0 ppm; 24: 205.9, 191.2 ppm), and a 13C NMR resonance at ca. 59 ppm (22: 58.8 ppm; 23: 58.2 ppm; 24: 59.1 ppm) that displays a bromine induced isotopic shift (for 22, Δδ = 1.6 ppb).‡ NOEs from H → R (22, NOESY; 23, 4.58%) confirm the bromide has shifted rather than a possible hydride shift (interchanging R and R′). However, their expected molecular ions with the characteristic bromine isotope pattern could not be observed by mass spectrometry, and only m/z 135 (Ad+) was observed by EI+ or CI+ methods (for 23, 24). |
| ††† The yields represent the optimized yields for the bromoallene oxides using 3.0 equivalents of DMDO where the total mass recoveries from these experiments are >90%. The use of 1.5 equivalents gave the bromoallene oxides 19–21 in 21%, 19% and 21% isolated yield respectively, with (for 7 and 8) 8% and 6% of 22 and 23, and recovered starting materials 7 (68%) and 8 (74%) for >95% total mass recovery. The use of 5.0 equivalents (for 7 and 8) led instead to the predominant formation of the corresponding bromodiketones 22 and 23 where no bromoallene oxides were observed. |
| ‡‡‡ These results are also congruent with the reported epoxidations of tri(alkyl)substituted allenes (ref. 4 and 10b) where (i) the most substituted alkene is epoxidised, and (ii) the epoxidation occurs with face selectivity for the resulting E-olefin. |
| §§§ A search of the Cambridge Structural Database (version 5.37, Feb-2016 update) for an allene oxide moiety returned only 3 hits, all of which are epoxyC60fullerenes. The O–C bond lengths in the epoxide moiety of bromoallene oxide 19 show a marked asymmetry. Whilst that to the sp3 carbon [O1–C1 1.453(6) Å] is close to the average C–O bond length in epoxides with two sp3 carbon centres [1.446 Å], that to the sp2 carbon of the adjacent CC double bond is significantly shorter [O1–C2 1.388(7) Å]. The C–C bond of the epoxide moiety [C1–C2 1.435(8) Å] is also shorter than the average for epoxides with two sp3 carbon centres [1.467 Å]. The C2–C3 double bond [1.304(8) Å] is unchanged from the average seen in allene systems [1.317 Å] (data for average bond lengths of CC double bonds in allene units, and in epoxides with two sp3 carbon centres came from statistical analyses of searches of the Cambridge Structural Database). |
| ¶¶¶ The question of the preferred site of epoxidation in 1-bromo-3-alkyl allenes (ref. 11) remains experimentally unresolved however. |
| This journal is © The Royal Society of Chemistry 2016 |