
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Morphology and phase transformation from NaCaSiO3OH to Na2Ca2Si2O7 and photoluminescence evolution via Eu3+/Tb3+ doping†
Mingyue
Chen
a,
Zhiguo
Xia
*a,
Maxim S.
Molokeev
bc and
Qiulin
Liu
a
aSchool of Materials Sciences and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: xiazg@ustb.edu.cn; Fax: +86-10-8237-7955; Tel: +86-10-8237-7955
bLaboratory of Crystal Physics, Kirensky Institute of Physics, SB RAS, Krasnoyarsk 660036, Russia
cDepartment of Physics, Far Eastern State Transport University, Khabarovsk, 680021, Russia
First published on 19th August 2016
Abstract
A facile and controllable ethanol/water aided hydrothermal process was developed to prepare the NaCaSiO3OH:Tb3+/Eu3+ phosphor. The morphologies were in situ constructed with the phase transformation from NaCaSiO3OH to Na2Ca2Si2O7, and the intrinsic crystal structural transformation mechanism and the dependence of their photoluminescence tuning on the Tb3+/Eu3+ ratio have been discussed.
The design and synthesis of rare earth (RE) ions doped luminescent materials with controllable morphologies remains a challenge owing to their size/shape-dependent properties and induced potential applications in lighting devices,1 biological labeling,2 lasers, displays,3etc. The physical–chemical properties of materials could be controlled as we desired through a good manipulation of the size and morphology.4 Therefore, fabrication of RE doped luminescent materials with controlled structures and morphologies is still one of the challenging issues in chemistry and materials science. Amongst them, RE doped silicate materials have been widely investigated due to the versatility in their crystal structures and tunable luminescence properties, which are suitable for application to many fields.5–8 Usually, high-temperature solid-state reactions are employed to prepare silicate materials. So, the resultant samples appear in aggregation with an irregular shape, and such a preparation method limits the possibility of tuning the morphology and the following properties of the final products. Accordingly, various solution-based preparation techniques, such as co-precipitation,9 hydrothermal,10 sol–gel,11 have been explored to prepare silicate materials with controllable morphologies, which is still a challenge.
Herein, we present a facile pathway toward Eu3+/Tb3+-doped Na2Ca2Si2O7 phosphors that rely on the heat treatment of the NaCaSiO3OH:Eu3+/Tb3+ precursor prepared using a controllable hydrothermal method. By tuning the ethanol/water (EtOH/H2O) ratio, it is possible to yield different precursor morphologies, such as rods, decahedron, distorted octahedron and spindle. Then, we synthesized Na2Ca2Si2O7 with the same particle shapes as those of NaCaSiO3OH, as obtained from the in situ construction and phase transformation from NaCaSiO3OH to Na2Ca2Si2O7. Additionally, the luminescence properties of Eu3+/Tb3+ doped NaCaSiO3OH and Na2Ca2Si2O7 microstructures are comparatively investigated, and the luminescent colors of Na2Ca2Si2O7:Tb3+,Eu3+ can be adjusted from red to green depending on the Tb3+/Eu3+ ratio, which should be ascribed to the energy transfer of Tb3+ → Eu3+. The work reveals that our current hydrothermal method following a heat treatment process is a facile and efficient way to the synthesis of such phosphors with controlled morphologies.
The preparation method employed for all the presented materials is described in detail in the ESI.† Herein, the X-ray diffraction (XRD) patterns of the NaCaSiO3OH samples prepared by the addition of different EtOH/H2O volume ratios are shown in Fig. 1. From Fig. 1 and Fig. S1a (ESI†), it can be observed that all samples exhibit a single phase and all peaks are assigned to the monoclinic phase NaCaSiO3OH (JCPDS card no. 25-1319). The dependence of the NaCaSiO3OH morphology on the reaction media of the EtOH/H2O volume ratio is illustrated well by the scanning electron microscopic (SEM) evaluation of the isolated products (see Fig. 2 and Fig. S1b, c, ESI†). Rod-like particles are obtained when the volume of H2O is 30 ml (Fig. 2a), and some rods are agglomerated to form rod bunches. From Fig. 2b, the morphology of NaCaSiO3OH is changing progressively to a nubby shape in the presence of 1 ml EtOH. Increasing the EtOH volume to 5 ml (EtOH/H2O = 5/25) yields a perfect decahedron shape noted in Fig. 2c, indicating that the decahedral structure may have grown from rods. Based on the high magnification SEM image (the inset of Fig. 2c), the decahedrons with some rips are highly uniform and symmetrical, and they exhibit a bigger size compared to the sample prepared in EtOH/H2O = 1/29. Upon addition of 10 ml EtOH, the decahedral shape transforms into octahedron one, but the two sides of the selected octahedron are not smooth (the inset of Fig. 2d). Upon further increasing the EtOH to 15 ml, the spindle shape is developed, and the centre thickness of the spindle is greater than the edge thickness. Then, under a rich amount of EtOH (20–30 ml), pure phase NaCaSiO3OH is still formed, but the morphology of NaCaSiO3OH particles become more irregular, even giving rise to crystal aggregates as illustrated in Fig. 2f and Fig. S1b, c (ESI†). More interestingly, it is observed that the higher the EtOH content (5–30 ml), the smaller the particle size. Therefore, we can conclude that the difference of morphologies and sizes in this surfactant/ligand-free system is mainly governed by the amount of the solvent EtOH. Generally, the growth of crystals and the formation of various morphologies are complex processes that are the cooperative results of the inherent structures and the external experimental conditions.12–14 In our case, two kinds of solvents (H2O and EtOH) with different polarities and saturated vapour pressures are used as reaction media, producing a distinguished morphology and a size of NaCaSiO3OH. The polarities and saturated vapour pressures of the solvents were found to affect the products under thermal conditions by giving adjustments to the homogenization of the reactants, the amount of individual nucleus formation, the amalgamation and direction preference of growing nucleus.15 Furthermore, the ratio of two solvents would affect the solubility of the starting materials, the reaction rate and the crystallization rate of products, which result in the various morphologies, as observed in the present and other experiments.
 | ||
| Fig. 1 XRD patterns of NaCaSiO3OH samples prepared in various volume ratios of EtOH to H2O. The standard data for NaCaSiO3OH (JCPDS card no. 25-1319) is shown as a reference. | ||
 | ||
| Fig. 2 SEM images of NaCaSiO3OH samples prepared in different volume ratios of EtOH to H2O: 0/30 (a), 1/29 (b), 5/25 (c), 10/20 (d), 15/15 (e) and 20/10 (f). The inset images in c–f are the corresponding enlarged single particle. | ||
The thermal stability of the NaCaSiO3OH phase is firstly investigated by thermogravimetric and differential scanning calorimetry (TG-DSC) analysis. From the recorded TG-DSC results during the heat-treatment of NaCaSiO3OH in air (Fig. 3a), it is apparent that the decomposition starts at 460 °C and finishes at 523 °C with the weight loss of 5.64%, which agrees with the theoretical value (5.76%) calculated from the dehydration of two NaCaSiO3OH molecules. Hence, the phase decomposition is proposed over the temperature range of 450–530 °C. To verify the occurrence of phase transformation and find the exact phase transformation temperature, variable temperature X-ray diffraction (VT-XRD) is employed in situ. As shown in VT-XRD patterns (Fig. 3b), we find that the NaCaSiO3OH phase is thermally stable up to 450 °C in air. At 500 °C, a mixture of the NaCaSiO3OH phase and the newly formed Na2Ca2Si2O7 phase (ICSD card no. 95858) appears. However, all the characteristic XRD peaks coincide with the standard pattern of Na2Ca2Si2O7, when the temperature is increased to 600 °C. Even if the heating temperature rises up to 1000 °C continuously, the Na2Ca2Si2O7 phase still maintained, confirming that the temperature of phase transformation from NaCaSiO3OH to Na2Ca2Si2O7 is about 500 °C, as observed from the TG-DSC results. Fig. 3c shows the SEM images of the resultant Na2Ca2Si2O7. More interestingly, it can be found that the morphologies of NaCaSiO3OH can be kept in situ even if it is sintered at 700 °C to form the Na2Ca2Si2O7 phase. We believe that such a phase transformation synthesis should be a general and facile way to prepare morphology-controllable silicate materials. Moreover, Fig. 3d gives the enlarged image of the individual Na2Ca2Si2O7 particle, and the porous surface character can be observed, which implies the decomposition of NaCaSiO3OH via evaporation of H2O molecular and the appearance of microscopic pores on the surface of the particles. The intrinsic crystal structural transformation mechanism will also be discussed in the following section.
 | ||
| Fig. 3 (a) TG-DSC curves of the as-obtained NaCaSiO3OH, (b) the VT-XRD patterns of NaCaSiO3OH precursors (EtOH/H2O = 5/25) by sintering them at different temperatures. (c) SEM images of resultant Na2Ca2Si2O7 when calcining the selected NaCaSiO3OH in situ at 700 °C and (d) the enlarged image of the single Na2Ca2Si2O7 particle. | ||
The phase transformation mechanism for the formation of the Na2Ca2Si2O7 phase with microscopic pores on the particle surface is proposed in Fig. 4. As we know, both the NaCaSiO3OH and Na2Ca2Si2O7 belong to monoclinic phases, but their space groups are P21/m and C2/c respectively and their cell parameters are absolutely distinguished. Therefore, such a phase transformation doesn't belong to the topotactic transformation because two structures have unit cells without relations. Besides these, other differences of the two structures are manifested as follows: (1) Na2Ca2Si2O7 consists of Ca, Na/Ca, Na and Si sites, but NaCaSiO3OH merely consists of Ca, Na and Si cations, no Ca/Na mixing exists in NaCaSiO3OH; (2) networks of SiO4 polyhedra are different, namely, there are isolated SiO4 in NaCaSiO3OH and two isolated groups (SiO4 and Si3O10) in Na2Ca2Si2O7 (see Fig. 4). However, the phase relations between NaCaSiO3OH and Na2Ca2Si2O7 should be considered. In this case we can suggest that OH groups of two SiO3OH tetrahedra combine with two H ions from other two SiO3OH tetrahedra forming two H2O molecules. The remaining two SiO3 and two SiO4 form Si3O10 + SiO4 as illustrated in the bottom panel of Fig. 4. The possible chemical reaction equation of the formation of Na2Ca2Si2O7 from NaCaSiO3OH could be described as follows:
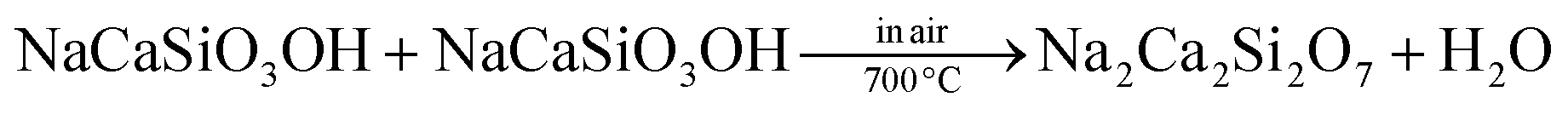
 | ||
| Fig. 4 The comparative description of the crystal structures of NaCaSiO3OH and Na2Ca2Si2O7, and the transformation mechanism from NaCaSiO3OH to Na2Ca2Si2O7. | ||
In the phase transition process, the dehydration from NaCaSiO3OH results in the generation of H2O; then, a large amount of vacancies should be formed during the formation of the new Na2Ca2Si2O7 phase. The expansion of vacancies may lead to the formation of cracks and microscopic pores at the Na2Ca2Si2O7 polyhedron facets. The SEM images of calcined product Na2Ca2Si2O7 (Fig. 3d) show that the polyhedrons possess many microscopic holes, which supports the above proposed mechanism.
Our experiments have also shown that both NaCaSiO3OH and Na2Ca2Si2O7 are good hosts for the luminescent materials formed via the Eu3+/Tb3+ doping. It should be mentioned that the doping of Eu3+/Tb3+ into NaCaSiO3OH and Na2Ca2Si2O7 does not change the phase purity and morphology of the final products as displayed in Fig. S3 (ESI†). Herein, the luminescence properties of Eu3+ and Tb3+ singly or/and codoped NaCaSiO3OH and Na2Ca2Si2O7 samples with a decahedron-like shape (EtOH/H2O = 5/25) were comparatively investigated. Fig. 5a and b show the photoluminescence (PL) emission spectra of NaCaSiO3OH:0.10Eu3+/0.03Tb3+ and Na2Ca2Si2O7:0.10Eu3+/0.03Tb3+ samples, respectively. NaCaSiO3OH:Eu3+ shows a strong red luminescence originating from the 5D0 → 7FJ (J = 0, 1, 2, 3, 4) transitions of Eu3+ ions under 254 nm excitation.16,17 For the Na2Ca2Si2O7:Eu3+ sample, the PL spectrum exclusively contains the characteristic emission of Eu3+ cantered at 611 nm. On the other hand, NaCaSiO3OH:Tb3+ shows a series of strong green emission lines with a maximum at about 542 nm, which are all ascribed to 5D4–7FJ (J = 6, 5, 4, 3) transitions of Tb3+ corresponding to 5D4–7F6 (488 nm), 5D4–7F5 (542 and 550 nm), 5D4–7F4 (583 nm) and 5D4–7F3 (625 nm), respectively, which are similar to other Tb3+-containing phosphors, such as Tb2(MoO4)3.18,19 For the Na2Ca2Si2O7:Tb3+, the emission spectral lines in the blue region are found to be at 416 nm (5D3–7F5) and 437 nm (5D3–7F4) with high emission intensity. However, they do not emerge in the emission spectra of NaCaSiO3OH:Tb3+, which means that there is a relatively low photon energy of Na2Ca2Si2O7 compared to that of NaCaSiO3OH. Moreover, the intensity ratio of 5D0 → 7F2 and 5D0 → 7F1 transitions of Eu3+ ions in different hosts is related to the activator symmetry occupied in the lattice. The strong emission from 5D0 → 7F2 compared to that of 5D0 → 7F1 in Na2Ca2Si2O7:0.10Eu3+ denotes that there is relatively low symmetry for Na2Ca2Si2O7. Therefore, it is found that there is obvious difference in the emission spectra of NaCaSiO3OH:Eu3+ and Na2Ca2Si2O7:Eu3+; as well as NaCaSiO3OH:Tb3+ and Na2Ca2Si2O7:Tb3+. The effects can be attributed to the different coordination environments and symmetry around the Tb3+/Eu3+ ions in the NaCaSiO3OH and Na2Ca2Si2O7 hosts.12 Furthermore, from Fig. 5c, the spectral overlap between the PL spectrum of Na2Ca2Si2O7:Tb3+ and photoluminescence excitation (PLE) spectrum of Na2Ca2Si2O7:Eu3+ can be observed, and the PL spectra of Na2Ca2Si2O7:Tb3+,Eu3+ consist of the peaks at 611 nm and 542 nm attributed to Eu3+ and Tb3+, respectively. The results suggest that the energy transfer can occur from Tb3+ to Eu3+,20 and the emitting colour can be tuned from green to yellow and red via energy transfer of Tb3+ → Eu3+ by tuning the doped Eu3+ concentrations as shown in Fig. 5d and the inset.
 | ||
| Fig. 5 PL spectra NaCaSiO3OH:0.10Eu3+/0.03Tb3+ (a) and Na2Ca2Si2O7:0.10Eu3+/0.03Tb3+ (b) phosphors. PLE and PL spectra of Na2Ca2Si2O7:0.03Eu3+ (c) -I, Na2Ca2Si2O7:0.03Tb3+ (c) -II, and Na2Ca2Si2O7:0.03Tb3+,0.03Eu3+ (c) -III phosphors. (d) PL spectra of Na2Ca2Si2O7:0.03Tb3+,xEu3+ phosphors with different Eu3+ doped concentrations (x), the inset shows the corresponding CIE chromaticity diagram. | ||
In order to further investigate the energy transfer process between Tb3+ and Eu3+, the decay curves of Tb3+ by monitoring the emission transition at 542 nm were measured and depicted in Fig. 6(a). The decay curve can be well fitted with a second-order exponential decay mode by using eqn (1):21–23
I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2) | (1) |
| τ* = (A1τ12 +A2τ22)/(A1τ1 + A2τ2) | (2) |
| ηT = 1 − (IS/IS0) = 1 − (τS/τS0) | (3) |
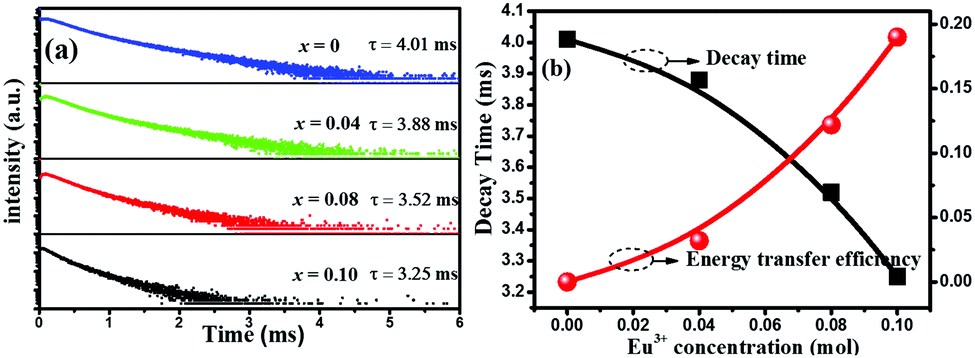 | ||
| Fig. 6 (a) The decay curves and lifetime values of Tb3+ in Na2Ca2Si2O7:0.03Tb3+,xEu3+ with different Eu3+ contents (x): x = 0, 0.04, 0.08 and 0.10. (b) Dependence of the fluorescence lifetime of the Tb3+ and energy transfer efficiency on doped Eu3+ molar concentration in Na2Ca2Si2O7:0.03Tb3+,xEu3+ samples. | ||
In summary, we have presented for the first time a simple hydrothermal method for the synthesis of highly crystallized NaCaSiO3OH particles with a controlled morphology/size in the presence of EtOH as both the solvent and the structure-directing agent. More importantly, the as-prepared NaCaSiO3OH can be in situ transferred into Na2Ca2Si2O7 by the heat-treatment in air, and the morphologies of the as-prepared NaCaSiO3OH have been kept without any agglomerations. In principle, this method offers a new alternative strategy to generate silicate materials. Moreover, when Na2Ca2Si2O7 is codoped with Eu3+ and Tb3+ with an appropriate ratio, the emission colours of the Na2Ca2Si2O7:Tb3+,Eu3+ phosphors can be tuned from green to yellow and red. It is expected that the morphology-controlled Na2Ca2Si2O7:Tb3+,Eu3+ micro-particles will provide potential applications for micro/nano functional devices.
The present work was supported by the National Natural Science Foundation of China (Grant No. 51572023 and 51272242), the Fundamental Research Funds for the Central Universities (FRF-TP-15-003A2) and the Russian Foundation for Basic Research (Grant No. 15-52-53080 GFEN_a).
Notes and references
- S. Sivakumar, F. C. J. M. van Veggel and M. Raudsepp, J. Am. Chem. Soc., 2005, 127, 12464 CrossRef CAS PubMed.
- G. S. Yi, H. C. Lu, S. Y. Zhao, G. Yue, W. J. Yang, D. P. Chen and L. H. Guo, Nano Lett., 2004, 4, 2191 CrossRef CAS.
- Phosphor Handbook, ed. S. Shionoya and W. M. Yen, CRC Press, Boca Raton, FL, 1999 Search PubMed.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- W.-W. Maria, M. Göckeritz, K. Uwe, L. Christoph and G. Gerald, Eur. J. Inorg. Chem., 2015, 2426 Search PubMed.
- Y. Wei, C. C. Lin, Z. W. Quan, M. S. Molokeev, V. V. Atuchin, T. S. Chan, Y. J. Liang, J. Lin and G. G. Li, RSC Adv., 2016, 6, 57261 RSC.
- H. P. Ji, L. Wang, M. S. Molokeev, N. Hirosaki, R. J. Xie, Z. H. Huang, Z. G. Xia, O. M. Ten Kate, L. H. Liu and V. V. Atuchin, J. Mater. Chem. C, 2016, 4, 6855 RSC.
- Z. G. Xia, Y. Y. Zhang, M. S. Molokeev, V. V. Atuchin and Y. Luo, Sci. Rep., 2013, 3, 3310 Search PubMed.
- J. S. Lee and Y. J. Kim, J. Nanosci. Nanotechnol., 2013, 13, 3685 CrossRef CAS PubMed.
- D. Ananias, F. A. A. Paz, D. S. Yufit, L. D. Carlos and J. Rochã, J. Am. Chem. Soc., 2015, 137, 3051 CrossRef CAS PubMed.
- R. Chrysafi, T. Perraki and G. Kakali, J. Eur. Ceram. Soc., 2007, 27, 1707 CrossRef CAS.
- G. G. Li, C. Peng, C. M. Zhang, Z. H. Xu, M. M. Shang, D. M. Yang, X. J. Kang, W. X. Wang, C. X. Li, Z. Y. Cheng and J. Lin, Inorg. Chem., 2010, 49, 10522 CrossRef CAS PubMed.
- C. V. Ramana, V. V. Atuchin, I. B. Troitskaia, S. A. Gromilov, V. G. Kostrovsky and G. B. Saupe, Solid State Commun., 2009, 149, 6 CrossRef CAS.
- V. V. Atuchin, T. A. Gavrilova, S. A. Gromilov, V. G. Kostrovsky, L. D. Pokrovsky, I. B. Troitskaia, R. S. Vemuri, G. C. Franco and C. V. Ramana, Cryst. Growth Des., 2009, 9, 1829 CAS.
- J. Zhang, L. D. Sun, J. L. Yin, H. L. Su, C. S. Liao and C. H. Yan, Chem. Mater., 2002, 14, 4172 CrossRef CAS.
- G. Jia, M. Yang, Y. H. Song, H. P. You and H. J. Zhang, Cryst. Growth Des., 2009, 9, 301 CAS.
- V. V. Atuchin, A. S. Aleksandrovsky, O. D. Chimitova, T. A. Gavrilova, A. S. Krylov, M. S. Molokeev, A. S. Oreshonkov, B. G. Bazarov and J. G. Bazarova, J. Phys. Chem. C, 2014, 118, 15404 CAS.
- Z. H. Xu, C. X. Li, G. G. Li, R. T. Chai, C. Peng, D. Y. Yang and J. Lin, J. Phys. Chem. C, 2010, 114, 2573 CAS.
- V. V. Atuchin, A. S. Aleksandrovsky, O. D. Chimitova, A. S. Krylov, M. S. Molokeev, B. G. Bazarov, J. G. Bazarova and Z. G. Xia, Opt. Mater., 2014, 36, 1631 CrossRef CAS.
- J. Zhou and Z. G. Xia, J. Mater. Chem. C, 2014, 2, 6978 RSC.
- C. H. Huang, T. M. Chen, W. R. Liu, Y. C. Chiu, Y. T. Yeh and S. M. Jang, ACS Appl. Mater. Interfaces, 2010, 2, 259 CAS.
- C. H. Huang and T. M. Chen, J. Phys. Chem. C, 2011, 115, 2349 CAS.
- G. Li, Y. Zhang, D. Geng, M. Shang, C. Peng, Z. Cheng and J. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 296 CAS.
- W. J. Yang and T. M. Chen, Appl. Phys. Lett., 2006, 88, 101903 CrossRef.
- K. H. Kwon, W. B. Im, H. S. Jang, H. S. Yoo and D. Y. Jeon, Inorg. Chem., 2009, 48, 11525 CrossRef CAS PubMed.
- P. I. Paulose, G. Jose, V. Thomas, N. V. Unnikrishnan and M. K. R. Warrier, J. Phys. Chem. Solids, 2003, 64, 841 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc06375f |
| This journal is © The Royal Society of Chemistry 2016 |