
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic quinolone signal analogues inhibiting the virulence factor elastase of Pseudomonas aeruginosa†
Dávid
Szamosvári
,
Valentin F.
Reichle
,
Monica
Jureschi
and
Thomas
Böttcher
 *
*
Department of Chemistry, Konstanz Research School Chemical Biology, University of Konstanz, 78457 Konstanz, Germany. E-mail: Thomas.Boettcher@uni-konstanz.de
First published on 26th September 2016
Abstract
We explore the chemical space of Pseudomonas quinolone signal analogs as privileged structures and report the discovery of a thioquinolone as a potent inhibitor of the important virulence factor elastase of the human pathogen Pseudomonas aeruginosa. We provide evidence that the derivative binds to the active site zinc of elastase and additionally acts as a fluorescent zinc sensor.
Pseudomonas aeruginosa is an important opportunistic human pathogen responsible for severe diseases ranging from urinary tract infections via life-threatening sepsis, endocarditis, and meningitis to chronic respiratory infections in cystic fibrosis patients.1,2 The increasing emergence of multi-drug resistant strains of P. aeruginosa poses major threats to public health and P. aeruginosa is one of the leading causes of hospital-acquired infections worldwide.3 The pathogenicity of P. aeruginosa is mediated by its enormous arsenal of virulence factors including toxins, extracellular enzymes, siderophores, and secretion systems that directly inject virulence factors into the eukaryotic host cell.4 A major virulence factor hereby is the enzyme elastase (LasB) that supports the infection and colonization process by damaging tissue and degrading immune proteins.5 The fine-tuned production of virulence factors that is responsible for the pathogen's broad spectrum of infective life-styles is orchestrated by the interactions of several intertwined quorum sensing systems.6–8 Inhibition of quorum sensing and its downstream circuits has attracted much attention as potential strategy to disarm pathogens for the future treatment of infectious diseases.9–12 One of the quorum sensing systems of P. aeruginosa, the Pseudomonas quinolone signalling (pqs) system, uses a series of 2-alkyl-4-quinolones (AQs) as signalling molecules.13 While various different AQs are known, the most abundant and well-studied are 3-hydroxy-2-heptyl-4-quinolone (PQS) and its biosynthetic precursor 2-heptyl-4-quinolone (HHQ) that may both have distinct roles in cell-to-cell communication.14,15 PQS and HHQ have been demonstrated to regulate virulence factor expression of P. aeruginosa and it has thus been suggested that targeting the pqs system may be a promising anti-virulence strategy.16,17 Furthermore, the HHQ and PQS quinolone scaffolds represent chemically privileged structures and we hence reasoned that heteroatom substituted derivatives may lead to functional diversity that could be applied to screen for potential virulence inhibitors. We thus synthesized a library of non-natural quinolone derivatives and report here the discovery of a potent inhibitor of the virulence factor elastase of pathogenic P. aeruginosa.
While previous studies have aimed to chemically inhibit or deregulate the pqs quorum sensing system using derivatives with modifications on the 2-alkyl-4-quinolone scaffold,18–20 we focussed on the synthetically more demanding approach of systematically changing the core scaffold by substituting its functional groups and replacing its heteroatoms. We thereby aimed to explore the chemical space of the privileged structures of HHQ and PQS-like non-quinolone compounds and investigate their biological activity as potential virulence inhibitors of P. aeruginosa. We first developed and evaluated various synthetic strategies towards the quinolone scaffold whereby we obtained PQS and HHQ as control compounds. HHQ (1) was prepared as reported previously by the synthesis of 3-oxodecanoic acid methyl ester, condensation with aniline and subsequent Conrad–Limpach cyclization (Fig. 1A).21 Although, HHQ was often used as starting point for the synthesis of 2-heptyl-3-hydroxyquinolin-4-one (PQS) by Duff-formylation and Dakin-oxidation as described by Pesci et al.,22 both reactions appeared to be problematic.21 Formylation of HHQ was only obtained when the HHQ was previously transferred into its quinoline tautomer. The following oxidation gave PQS in only 23% yield. PQS was therefore synthesized after the method of Hradil et al. which turned out to be a much more reliable and up scalable approach to prepare PQS (Fig. 1B).23,24
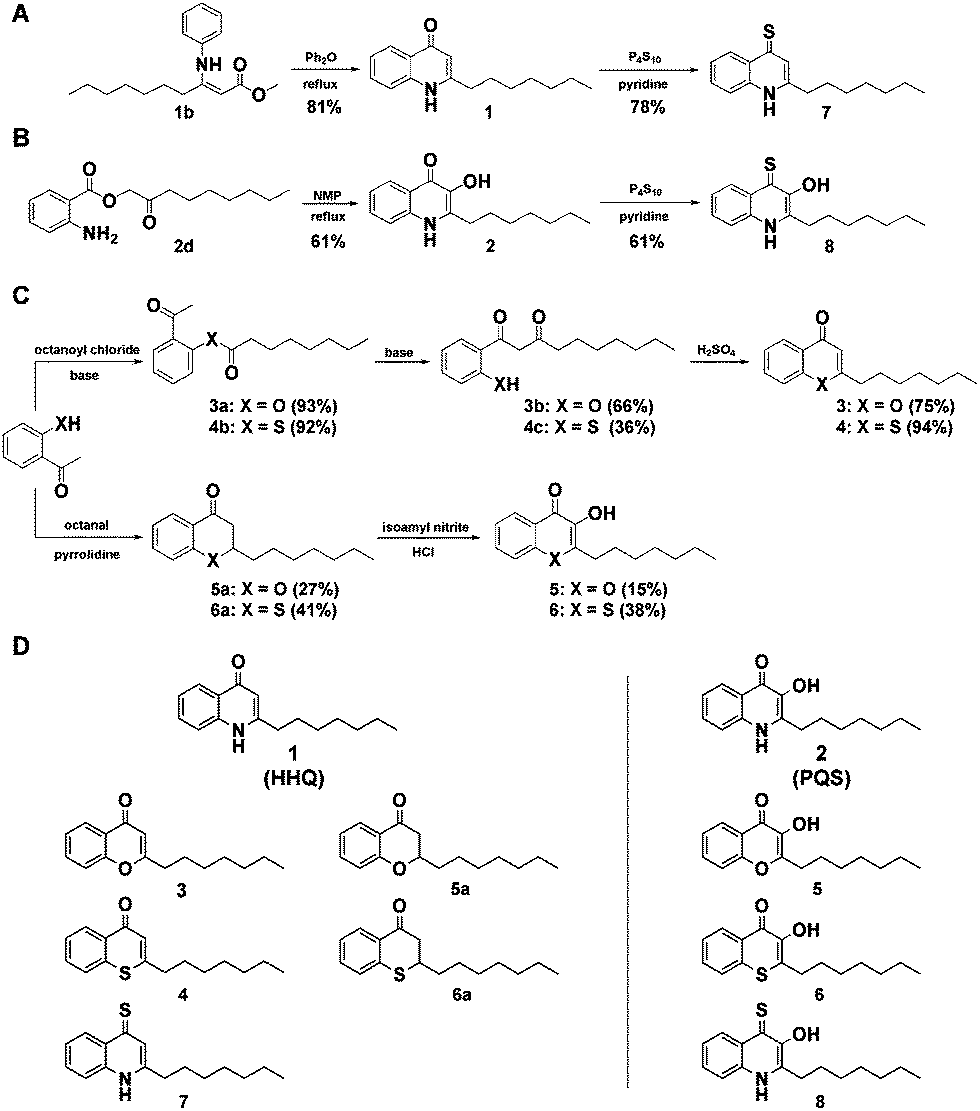 | ||
| Fig. 1 Synthetic library of HHQ and PQS derivatives. (A) Synthesis scheme for HHQ (1) and the derived thioketone 7, and (B) for PQS (2) and its analog 8. (C) Synthesis of compounds 3–6. (D) Structures of the compound library tested in the bioassays. | ||
To generate a structurally diverse library of HHQ and PQS derivatives, heteroatom substitutions were intended at positions 1, 3, and 4 of the 2-alkyl-4-quinolone scaffold. 4-Thioketo-analoges 7 and 8 were synthesized by thionation of the appropriate 4-keto-compounds HHQ (1) and PQS (2) using P4S10 in pyridine under reflux conditions (Fig. 1A and B).25 This reaction was found to be extremely reliable giving the desired products in good yields without interfering with hydroxyl- or amine functionalities at the same time whereas the Lawessons-reagent did not result successful thionation of the ketones.
Chromen-4-one (3) and thiochromen-4-one (4) were synthesized from their corresponding 1-3-diketones 3b and 4c which were prepared by Baker–Venkataraman rearrangement of the octanoyloxy esters of 2-hydroxyacetophenone and 2-mercaptoacetophenone 4a, respectively.26,27 Synthesis of the 1-O and 1-S-PQS-derivatives 5 and 6 started from 2-hydroxy- and 2-mercaptoacetophenone, respectively via the corresponding chroman-4-one 5a and thiochroman-4-one 6a by pyrrolidine catalyzed Knoevenagel-reaction with octanal (Fig. 1C).28,29 Oxidation of the α-keto position turned out to be difficult since an oxidation of the sulfide group to a sulfoxide or sulfone had to be avoided. The product was obtained by nitrosation with isoamyl nitrite and subsequent oximation and oxime hydrolysis.29 The combination of these diverse synthetic strategies resulted in a small library of 10 compounds (Fig. 1D). In order to investigate our library for potential biological activity we performed a series of virulence assays with live cells of the highly virulent P. aeruginosa strain PA14.30 We screened the library for inhibition of three important extracellular virulence factors, pyocyanine, rhamnolipid, and elastase. Cultures of P. aeruginosa PA14 were grown in liquid medium supplemented with 500 μM of each compound and after incubation for 24 h the production or activities of the corresponding virulence factors were quantified in spent culture supernatants.
While most compounds did not result in significant changes or slightly increased elastolytic activity (2 and 6), one compound (8) almost completely inhibited elastin degradation at a concentration of 500 μM (Fig. 2A and Fig. S1, S2, ESI†). The active compound did not inhibit growth of P. aeruginosa at the maximum tested concentration of 1 mM indicating that the effect on was not an artefact of cell toxicity or reduced growth (Fig. S3, ESI†). Elastolytic activity in cultures of P. aeruginosa is mainly caused by the extracellular virulence factor LasB (elastase), a zinc metalloprotease that contributes as major virulence factor to the infectious lifestyle of P. aeruginosa.31 As no other virulence factors tested were impacted, we speculated that coincidently 8 may inhibit directly the activity of the enzyme elastase rather than its production via the pqs quorum sensing system. To test this hypothesis we used purified elastase and employed an activity assay with a fluorogenic peptide substrate. This in vitro assay resulted in a very potent inhibition by 8 with an IC50 of 4 μM, confirming the direct mode of inhibition of elastolytic activity on enzyme level (Fig. 2B).
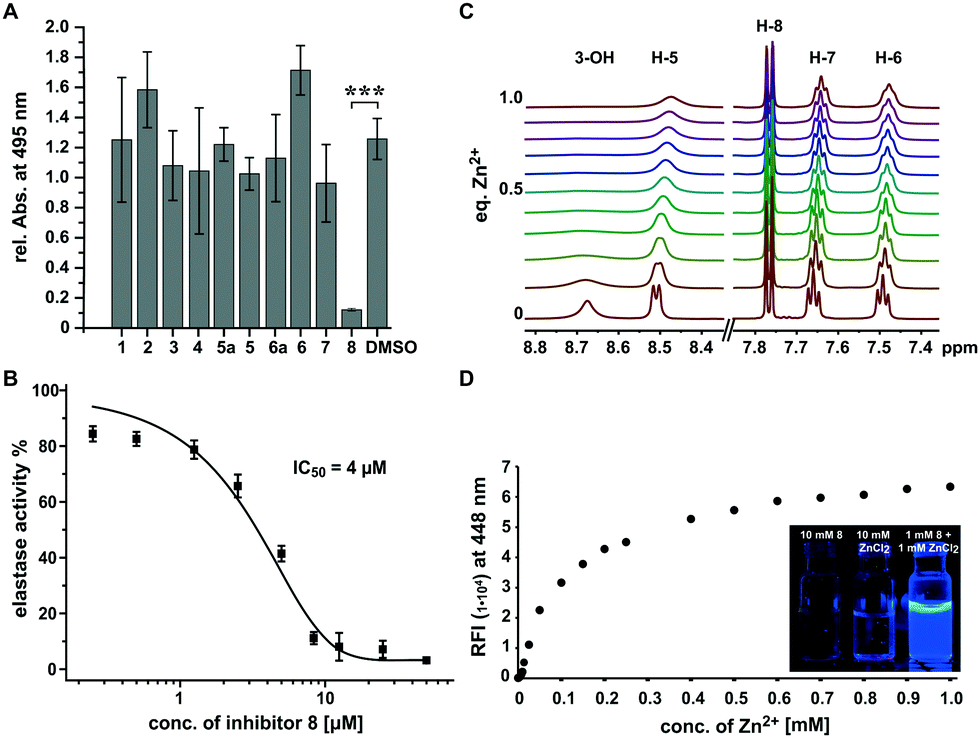 | ||
| Fig. 2 Active compound screening and investigations on the mechanism of 8. (A) Screening of the compound library for the effect on elastin–congo red degradation with DMSO as control. (B) In vitro inhibition of elastase activity by 8. (C) 1H NMR shifts of 8 in dependence of equivalents of added zinc(II). (D) Relative fluorescence increase of 8 in dependence of the zinc(II) concentration. Inset: Fluorescence of 8 under excitation by UV light at 365 nm. *** Independent two-sample t-test p < 0.00001. | ||
It is known that analogous structures of 8 bind to zinc and are metalloproteinase inhibitors and PQS is known as iron chelator.32–35 We thus speculated that the mechanism of action of 8 may involve binding to the zinc ion in the active site of elastase whereby its activity is inhibited. To investigate if 8 directly binds to zinc(II), we applied a combination of spectroscopic and NMR-based methods. NMR-titration of 8 with zinc chloride in DMSO resulted in significant shifts and broadening of 1H and 13C signals in dependence of the zinc(II) concentration (Fig. 2C and Fig. S4, S5, ESI†). Surprisingly, when zinc(II) was added to a solution of 8 in ethanol, a strong fluorescence was observed and a titration experiment revealed that zinc concentrations down to 2.5 μM could still be detected by fluorescence intensities two-fold over baseline making the compound also a formidable zinc sensor (Fig. 2D). In contrast, no fluorescence was observed for other biologically relevant divalent cations, confirming its high selectivity for zinc. These results suggest that 8 inhibits elastase by binding to the zinc ion in active site of this metalloenzyme.
In order to further elucidate the influence of heteroatoms in position 1 for elastase inhibition, we synthesized a second generation of analogues of 8 including derivatives with the nitrogen in position 1 of the 3-hydroxy-4-thioquinolone scaffold replaced by oxygen 9 and sulphur 10. Additionally we generated a 3-hydroxy-4-oxime derivative as an alternative metal chelator (11). The 4-thioketones 9 and 10 were synthesized by thionation with P4S10 in pyridine as described for 7 and 8 (Fig. 3A). Interestingly, the oxime 11 could not be obtained by base catalyzed reaction of PQS with hydroxylamine hydrochloride which is probably because the keto-form can be also understood as vinylogous amide36 instead, 3-hydroxyl TBDMS protected PQS was transferred into its enol-tautomer and 4-hydroxyl benzylated to allow the oxime formation at position 4 (Fig. 3B).
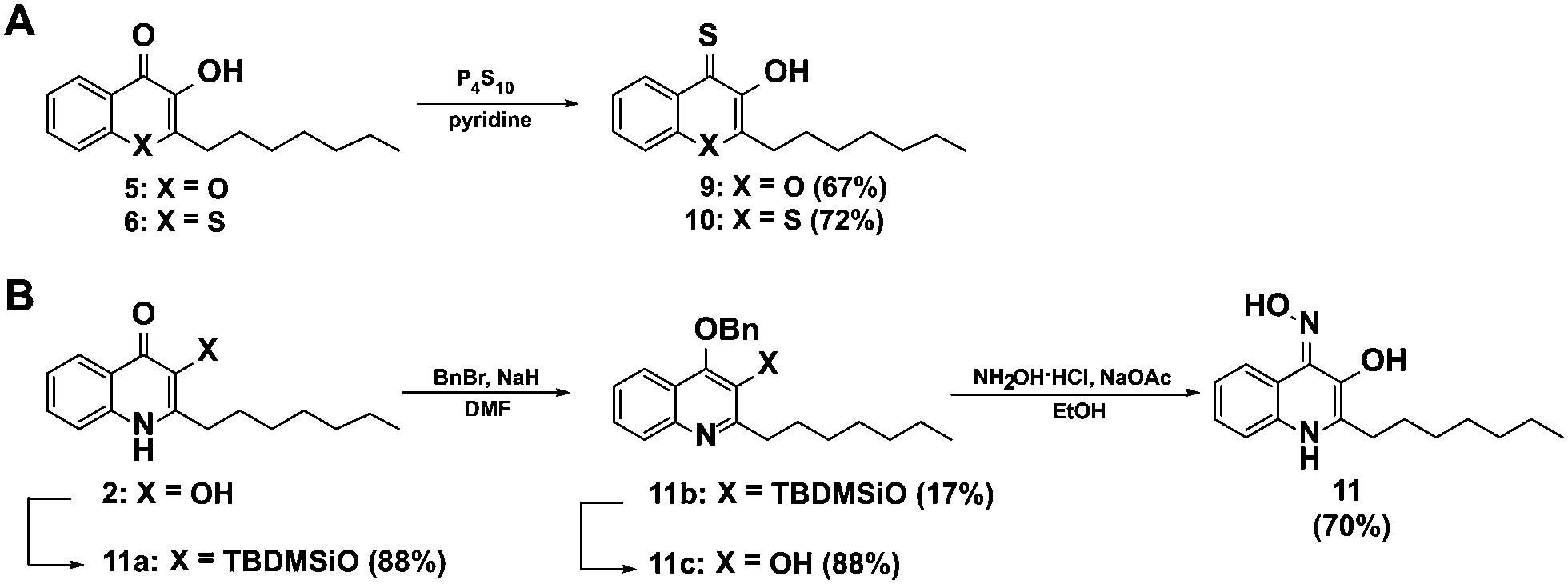 | ||
| Fig. 3 Synthesis of a second generation of compounds based on the active elastase inhibitor 8. (A) Scheme for the synthesis of the thioquinolone derivatives 9 and 10, and (B) for the oxime 11. | ||
Using this set of compounds, we first investigated their ability to inhibit elastase activity in vitro. Hereby, compounds 9 and 10 were even slightly more active than 8, each with an IC50 of 2 μM (Fig. 4A and Fig. S6, ESI†). In contrast, compound 11 did not inhibit elastase activity in vitro at concentrations up to 50 μM indicating that sulphur in position 4 is required for activity of the compound and cannot be simply replaced by other chelating groups (Fig. S7, ESI†). While compounds 9 and 10 did not exhibit fluorescence in presence of zinc, a competitive spectroscopic experiment with 8 as zinc sensor allowed to detect fluorescence quenching at increasing concentrations of 9 and 10, indicating that these compounds competed with 8 for zinc binding (Fig. S8, ESI†). Consequently, the inhibition of elastase involved most likely for all three compounds the binding of a hydroxyl thioketone or its corresponding thioenol-form to the active site zinc. For the in vitro studies the higher activity of the electron deficient aromatic systems of compounds 9 and 10 first appeared to be puzzling. However, analysis of the crystal structure of elastase revealed a carboxyl group (Glu141) in proximity to the active site which might stabilize the positive charge in the thioenol-form (Fig. 4B). Hydrophobic pockets in proximity to the active site appear to be ideal for accommodating the lipophilic heptyl chain of our compounds and also may explain the preference for hydrophobic side groups in natural substrates and previously reported elastase inhibitors.34,37,38
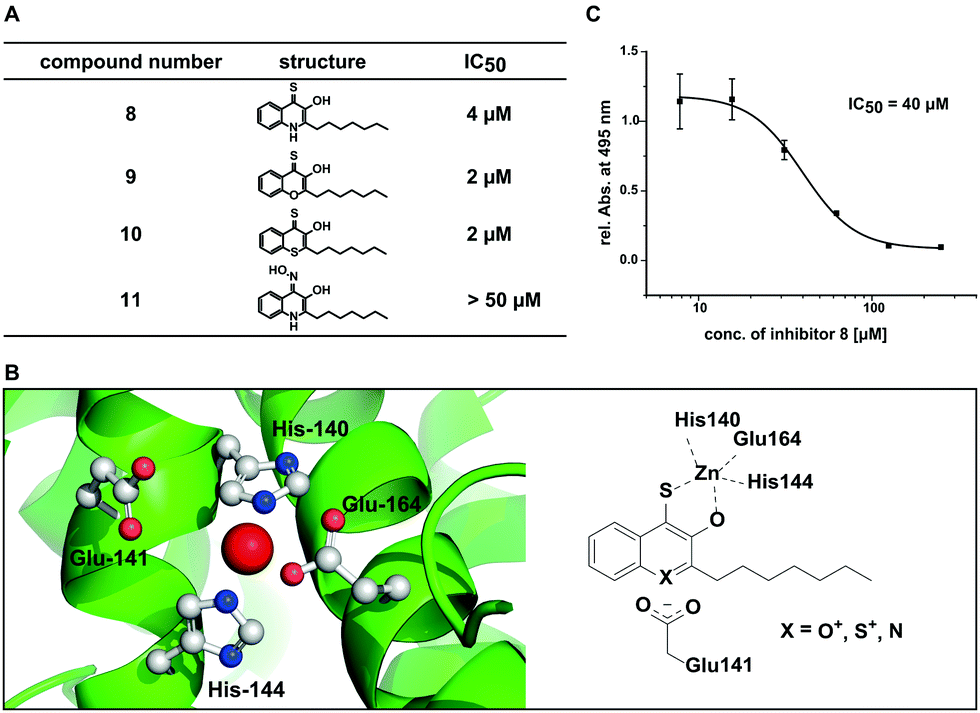 | ||
| Fig. 4 Inhibition of Pseudomonas elastase activity. (A) Activity of the second generation of compounds derived from 8 with elastase in vitro. (B) Active site of LasB with His140, His144, and Glu164 coordinating the zinc ion and proposed mechanism of inhibition. (C) In situ inhibition of elastolytic activity with compound 8 in culture of P. aeruginosa PA14. | ||
To quantify the in situ efficacy, we measured the concentration dependent inhibition of elastolytic activity by compounds 8–11 with live cultures of P. aeruginosa PA14. Hereby, only compound 8 was an efficient inhibitor of elastolytic activity in situ resulting in sigmoidal inhibition behaviour with an IC50 of 40 μM (Fig. 4C). Compound 10 resulted in only low efficacy with an in situ IC50 of 351 μM and compounds 9 and 11 were inactive up to 500 μM (Fig. S9, ESI†). The discrepancy between in vitro and in situ activity of 9 and 10 may be explained by their similarity to flavones that are known to be degraded by Pseudomonads.39 We thus suspect that the compounds may be fed into bacterial metabolism reducing their half-life and thus their efficacy whereby the magnitude of the drop in activity from in vitro to in situ experiments correlates with the increasing similarity of the compounds with the flavone scaffold. With 8 being the most active compound in situ we have discovered a potent inhibitor of Elastase (LasB) as major virulence factor of P. aeruginosa PA14 that is responsible for the pathogen's ability to evade the immune response and establish life-threatening infections.40,41
In conclusion, PQS derived quinolones with heteroatom substitutions represent highly interesting privileged structures that can be easily accessed by organic synthesis. Specifically, we demonstrate that 3-hydroxy-4-thioquinolone derivatives are promising candidates for the development of customized elastase inhibitors. We show evidence that our most active compound binds directly to the active site zinc of the enzyme and inhibits elastolytic activity in vitro and also in cultures of live cells. Our newly developed core scaffold thus represents an unprecedented chemical tool for studying elastase function and highly promising lead structure for further development of potential anti-virulence drugs.
We thank Prof. Andreas Marx and his group for their generous support. We gratefully acknowledge funding by the Emmy Noether program of the Deutsche Forschungsgemeinschaft (DFG), EU FP7 Marie Curie Zukunftskolleg Incoming Fellowship Program – University of Konstanz grant no. 291784, Fonds der Chemischen Industrie (FCI), Konstanz Research School Chemical Biology (KoRS-CB), and CRC969 (DFG). DS was supported by a KoRS-CB PhD fellowship. We thank PD Dr David Schleheck and Prof. Christof Hauck for the use of their S2 facilities, and Atul Pawar for help with Pymol.
Notes and references
- A. Oliver, R. Canton, P. Campo, F. Baquero and J. Blazquez, Science, 2000, 288, 1251–1254 CrossRef CAS PubMed.
- G. P. Bodey, R. Bolivar, V. Fainstein and L. Jadeja, Rev. Infect. Dis., 1983, 5, 279–313 CrossRef CAS PubMed.
- V. Aloush, S. Navon-Venezia, Y. Seigman-Igra, S. Cabili and Y. Carmeli, Antimicrob. Agents Chemother., 2006, 50, 43–48 CrossRef CAS PubMed.
- T. Strateva and I. Mitov, Ann. Microbiol., 2011, 61, 717–732 CrossRef CAS.
- B. Wretlind and O. R. Pavlovskis, Rev. Infect. Dis., 1983, 5(suppl. 5), S998–S1004 CrossRef CAS PubMed.
- R. S. Smith and B. H. Iglewski, Curr. Opin. Microbiol., 2003, 6, 56–60 CrossRef CAS PubMed.
- M. Schuster and E. P. Greenberg, Int. J. Med. Microbiol., 2006, 296, 73–81 CrossRef CAS PubMed.
- M. A. Welsh and H. E. Blackwell, Cell Chem. Biol., 2016, 23, 361–369 CrossRef CAS PubMed.
- M. A. Welsh, N. R. Eibergen, J. D. Moore and H. E. Blackwell, J. Am. Chem. Soc., 2015, 137, 1510–1519 CrossRef CAS PubMed.
- Q. H. Christensen, T. L. Grove, S. J. Booker and E. P. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13815–13820 CrossRef CAS PubMed.
- T. Böttcher and S. A. Sieber, J. Am. Chem. Soc., 2008, 130, 14400–14401 CrossRef PubMed.
- J. D. Moore, F. M. Rossi, M. A. Welsh, K. E. Nyffeler and H. E. Blackwell, J. Am. Chem. Soc., 2015, 137, 14626–14639 CrossRef CAS PubMed.
- S. McGrath, D. S. Wade and E. C. Pesci, FEMS Microbiol. Lett., 2004, 230, 27–34 CrossRef CAS PubMed.
- E. Deziel, F. Lepine, S. Milot, J. He, M. N. Mindrinos, R. G. Tompkins and L. G. Rahme, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1339–1344 CrossRef CAS PubMed.
- H. Huse and M. Whiteley, Chem. Rev., 2011, 111, 152–159 CrossRef CAS PubMed.
- J. F. Dubern and S. P. Diggle, Mol. BioSyst., 2008, 4, 882–888 RSC.
- M. W. Calfee, J. P. Coleman and E. C. Pesci, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11633–11637 CrossRef CAS PubMed.
- M. Starkey, F. Lepine, D. Maura, A. Bandyopadhaya, B. Lesic, J. X. He, T. Kitao, V. Righi, S. Milot, A. Tzika and L. Rahme, PLoS Pathog., 2014, 10, e1004321 Search PubMed.
- C. Lu, B. Kirsch, C. K. Maurer, J. C. de Jong, A. Braunshausen, A. Steinbach and R. W. Hartmann, Eur. J. Med. Chem., 2014, 79, 173–183 CrossRef CAS PubMed.
- A. Ilangovan, M. Fletcher, G. Rampioni, C. Pustelny, K. Rumbaugh, S. Heeb, M. Camara, A. Truman, S. R. Chhabra, J. Emsley and P. Williams, PLoS Pathog., 2013, 9, e1003508 CAS.
- F. J. Reen, S. L. Clarke, C. Legendre, C. M. McSweeney, K. S. Eccles, S. E. Lawrence, F. O'Gara and G. P. McGlacken, Org. Biomol. Chem., 2012, 10, 8903–8910 CAS.
- E. C. Pesci, J. B. Milbank, J. P. Pearson, S. McKnight, A. S. Kende, E. P. Greenberg and B. H. Iglewski, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11229–11234 CrossRef CAS.
- P. Hradil, J. Hlavac and K. Lemr, J. Heterocycl. Chem., 1999, 36, 141–144 CrossRef CAS.
- J. T. Hodgkinson, W. R. J. D. Galloway, S. Saraf, I. R. Baxendale, S. V. Ley, M. Ladlow, M. Welch and D. R. Spring, Org. Biomol. Chem., 2011, 9, 57–61 CAS.
- J. Bergman, B. Pettersson, V. Hasimbegovic and P. H. Svensson, J. Org. Chem., 2011, 76, 1546–1553 CrossRef CAS PubMed.
- P. Nanjan, J. Nambiar, B. G. Nair and A. Banerji, Bioorg. Med. Chem., 2015, 23, 3781–3787 CrossRef CAS PubMed.
- J. I. Lee and M. J. Kim, Bull. Korean Chem. Soc., 2011, 32, 1383–1386 CrossRef CAS.
- H. J. Kabbe and A. Widdig, Angew. Chem., Int. Ed., 1982, 21, 247–256 CrossRef.
- M. Ferrali, S. Bambagioni, A. Ceccanti, D. Donati, G. Giorgi, M. Fontani, F. Laschi, P. Zanello, M. Casolaro and A. Pietrangelo, J. Med. Chem., 2002, 45, 5776–5785 CrossRef CAS PubMed.
- H. Mikkelsen, R. McMullan and A. Filloux, PLoS One, 2011, 6, e29113 CAS.
- D. R. Galloway, Mol. Microbiol., 1991, 5, 2315–2321 CrossRef CAS PubMed.
- S. Johnson, E. Barile, B. Farina, A. Purves, J. Wei, L. H. Chen, S. Shiryaev, Z. Zhang, I. Rodionova, A. Agrawal, S. M. Cohen, A. Osterman, A. Strongin and M. Pellecchia, Chem. Biol. Drug Des., 2011, 78, 211–223 CAS.
- A. Agrawal, S. L. Johnson, J. A. Jacobsen, M. T. Miller, L. H. Chen, M. Pellecchia and S. M. Cohen, ChemMedChem, 2010, 5, 195–199 CrossRef CAS PubMed.
- A. L. Garner, A. K. Struss, J. L. Fullagar, A. Agrawal, A. Y. Moreno, S. M. Cohen and K. D. Janda, ACS Med. Chem. Lett., 2012, 3, 668–672 CrossRef CAS PubMed.
- S. P. Diggle, S. Matthijs, V. J. Wright, M. P. Fletcher, S. R. Chhabra, I. L. Lamont, X. Kong, R. C. Hider, P. Cornelis, M. Camara and P. Williams, Chem. Biol., 2007, 14, 87–96 CrossRef CAS PubMed.
- S. Hibino, E. Sugino, T. Choshi and K. Sato, J. Chem. Soc., Perkin Trans. 1, 1988, 2429–2432 RSC.
- G. R. Cathcart, D. Quinn, B. Greer, P. Harriott, J. F. Lynas, B. F. Gilmore and B. Walker, Antimicrob. Agents Chemother., 2011, 55, 2670–2678 CrossRef CAS PubMed.
- N. Nishino and J. C. Powers, J. Biol. Chem., 1980, 255, 3482–3486 CAS.
- B. V. Pillai and S. Swarup, Appl. Environ. Microbiol., 2002, 68, 143–151 CrossRef CAS PubMed.
- M. J. van der Plas, R. K. Bhongir, S. Kjellstrom, H. Siller, G. Kasetty, M. Morgelin and A. Schmidtchen, Nat. Commun., 2016, 7, 11567 CrossRef PubMed.
- Z. Kuang, Y. Hao, B. E. Walling, J. L. Jeffries, D. E. Ohman and G. W. Lau, PLoS One, 2011, 6, e27091 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc06295d |
| This journal is © The Royal Society of Chemistry 2016 |