
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free C–H sulfonamidation of pyrroles by visible light photoredox catalysis†
Andreas Uwe
Meyer‡
,
Anna Lucia
Berger‡
and
Burkhard
König
 *
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstrasse 31, 93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de; Fax: (+49)-941-943-1717; Tel: (+49)-941-943-4575
First published on 12th August 2016
Abstract
We report a one-step procedure for the preparation of N-(2-pyrrole)-sulfonamides from sulfonamides and pyrroles. The reaction uses visible light, an acridinium dye as photocatalyst and oxygen as the terminal oxidant for the oxidative C–N bond formation; structures of several reaction products were confirmed by X-ray structure analysis. The reaction is selective for pyrroles, due to the available oxidation power of the photocatalyst and the required stability of the carbocation intermediate under the reaction conditions.
Sulfonamides are an important class of organic compounds1,2 finding applications in medicinal chemistry e.g. as Janus kinase (JAK) inhibitors for treating psoriasis and other inflammatory skin disorders,3 HCV NS5B polymerase inhibitors for the treatment of hepatitis C virus,4 and GPAT (glycerol 3-phosphate acyltransferase) inhibitors.5 The heterocyclic sulfonamide drug rosuvastatin (Fig. 1), a HMG-CoA reductase inhibitor, was among the worldwide most sold pharmaceuticals in 20116 and 2013.7
 | ||
| Fig. 1 The sulfonamide drug rosuvastatin. | ||
Typical C–H sulfonamidation methods require transition metals8,9 as for example the palladium-catalyzed intermolecular coupling of aryl chlorides and sulfonamides under microwave irradiation (Scheme 1a),10 the palladium-catalyzed intramolecular sulfonamidation of imines,11 the iridium-catalyzed reactions of arenes12,13 and heteroarenes with sulfonyl azides (Scheme 1b),14 and the copper-mediated C–H sulfonamidation, which requires stoichiometric amounts of copper.15 Another approach is the sulfonamidation of indoles by stoichiometric amounts of iodine.16–18 In 2008 Moeller et al. developed an intramolecular sulfonamidation of alkenes by electrochemical oxidation of electron-rich double bonds and subsequent C–N bond formation with a sulfonamide anion19–22 and in 2013 Nicewicz et al. published the catalytic anti-Markovnikov intramolecular hydroamination following a similar concept. They oxidized the alkene with the organic photocatalyst 9-mesityl-10-methylacridinium23 followed by an intramolecular reaction with the sulfonamide.24–26 Recently, Nicewicz and co-workers extended this method to a site-selective C–H amination by oxidizing electron-rich aromatics with an acridinium dye and subsequent reaction with amines.27
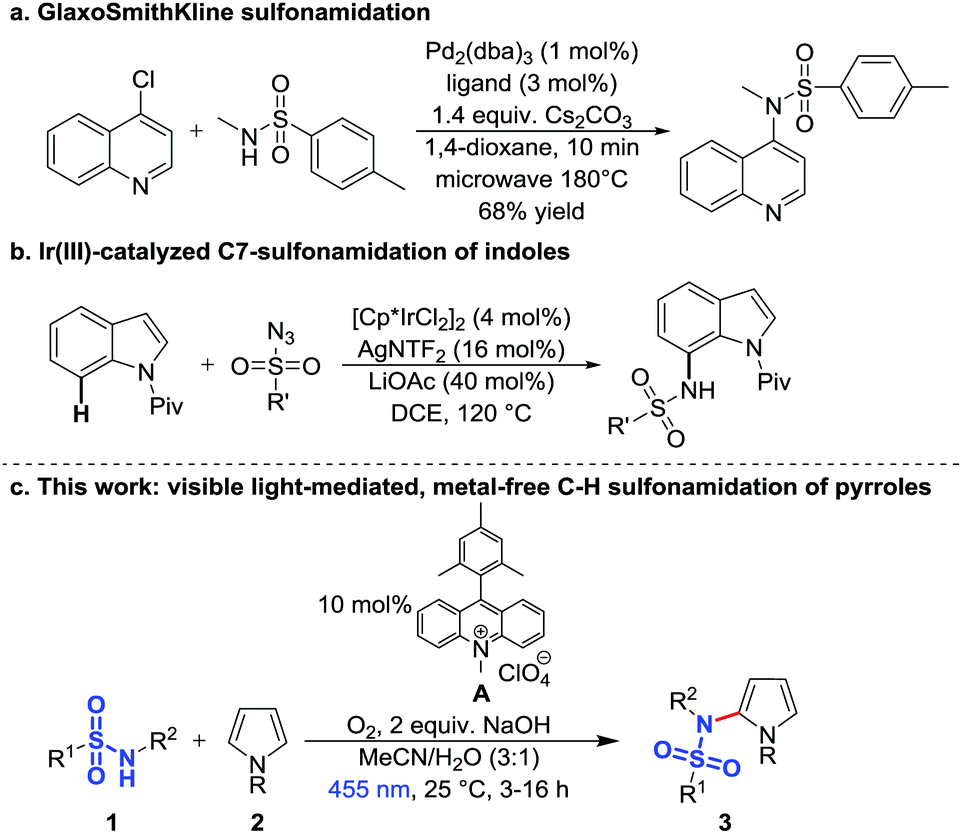 | ||
| Scheme 1 Transition metal-catalyzed and photocatalytic reactions for C–H sulfonamidations. | ||
However, for the synthesis of many drug motifs an intermolecular sulfonamidation of heteroarenes would be useful. A particular interesting target structure in this respect is pyrrole due to its presence in many biologically important compounds28–33 and active drugs, e.g. atorvastatin,34 one of the best-selling drugs of the last years.32,35,36 Based on previous results we have therefore developed a metal-free photocatalytic C–H sulfonamidation of pyrroles using blue light, the commercially available organic dye 9-mesityl-10-methylacridinium perchlorate (A) as photocatalyst, oxygen as the terminal oxidant and sodium hydroxide as base (Scheme 1c).
The reaction conditions were optimized by irradiating a mixture of N-ethyl-4-methylbenzene-1-sulfonamide (1a), N-methyl-pyrrole (2a), 9-mesityl-10-methylacridinium perchlorate (A), sodium hydroxide and oxygen with blue light at room temperature. Different catalysts, bases, oxidants, varying amounts of trapping reagent and different irradiation times were investigated (Table 1).
|
|
||
|---|---|---|
| Entry | Conditions | Yielda [%] |
| a Determined by GC analysis with naphthalene as internal standard. | ||
| 1 | A (10 mol%), n = 20, x = 2, 3 h | 98 |
| 2 | A (10 mol%), n = 20, x = 2, 6 h | 99 |
| 3 | A (10 mol%), n = 20, x = 2 | 99 |
| 4 | No photocatalyst, n = 20, x = 2 | — |
| 5 | A (10 mol%), n = 20, x = 2, no light | — |
| 6 | No photocatalyst, n = 20, x = 2, no light | — |
| 7 | A (10 mol%), n = 20, x = 2, no base | — |
| 8 | A (10 mol%), n = 20, x = 2, no oxidant | 16 |
| 9 | A (10 mol%), n = 10, x = 2 | 99 |
| 10 | A (10 mol%), n = 5, x = 2 | 95 |
| 11 | A (5 mol%), n = 20, x = 2 | 83 |
| 12 | A (10 mol%), n = 20, x = 1 | 40 |

In a typical reaction mixture for the photocatalytic reaction, one equivalent of the sulfonamide 1a, 20 equivalents of N-Me-pyrrole (2a), two equivalents of sodium hydroxide and 10 mol% of 9-mesityl-10-methylacridinium perchlorate (A) in a mixture of MeCN/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with an oxygen-balloon were used to give 3a in 98% yield for 3 h and 99% yield for 6 and 16 hours, respectively, (Table 1, entries 1–3). The catalyst with tetrafluoroborate as counter ion is working equally good in the reaction. Control experiments without photocatalyst, light or base, confirmed that all components are necessary for product formation (Table 1, entries 4–7). Without oxygen balloon the yield dropped to 16% (Table 1, entry 8). The excess of the heteroarene 2a can be reduced to 10 equiv. with still quantitative product yields and 5 equiv. giving 95% yield (Table 1, entries 9 and 10). A catalyst loading of 5 mol% gave 83% of 3a and just one equivalent of sodium hydroxide decreased the yield to 40% (Table 1, entries 11 and 12). Nitrobenzene (18%) and ammonium persulfate are not suitable oxidants (see ESI,† Table S1, entries 1 and 2). The bases potassium hydroxide (45%), potassium phosphate (36%), potassium tert-butoxide (31%) and the weak bases potassium carbonate (13%), cesium carbonate (6%) and cesium fluoride (no product formation) are less efficient (see ESI,† Table S1, entries 3–8). Other photocatalysts like Ru(bpy)3Cl237 and eosin Y38–40 were tried for the oxidation under various conditions, but no product formation occurred.
:1) with an oxygen-balloon were used to give 3a in 98% yield for 3 h and 99% yield for 6 and 16 hours, respectively, (Table 1, entries 1–3). The catalyst with tetrafluoroborate as counter ion is working equally good in the reaction. Control experiments without photocatalyst, light or base, confirmed that all components are necessary for product formation (Table 1, entries 4–7). Without oxygen balloon the yield dropped to 16% (Table 1, entry 8). The excess of the heteroarene 2a can be reduced to 10 equiv. with still quantitative product yields and 5 equiv. giving 95% yield (Table 1, entries 9 and 10). A catalyst loading of 5 mol% gave 83% of 3a and just one equivalent of sodium hydroxide decreased the yield to 40% (Table 1, entries 11 and 12). Nitrobenzene (18%) and ammonium persulfate are not suitable oxidants (see ESI,† Table S1, entries 1 and 2). The bases potassium hydroxide (45%), potassium phosphate (36%), potassium tert-butoxide (31%) and the weak bases potassium carbonate (13%), cesium carbonate (6%) and cesium fluoride (no product formation) are less efficient (see ESI,† Table S1, entries 3–8). Other photocatalysts like Ru(bpy)3Cl237 and eosin Y38–40 were tried for the oxidation under various conditions, but no product formation occurred.
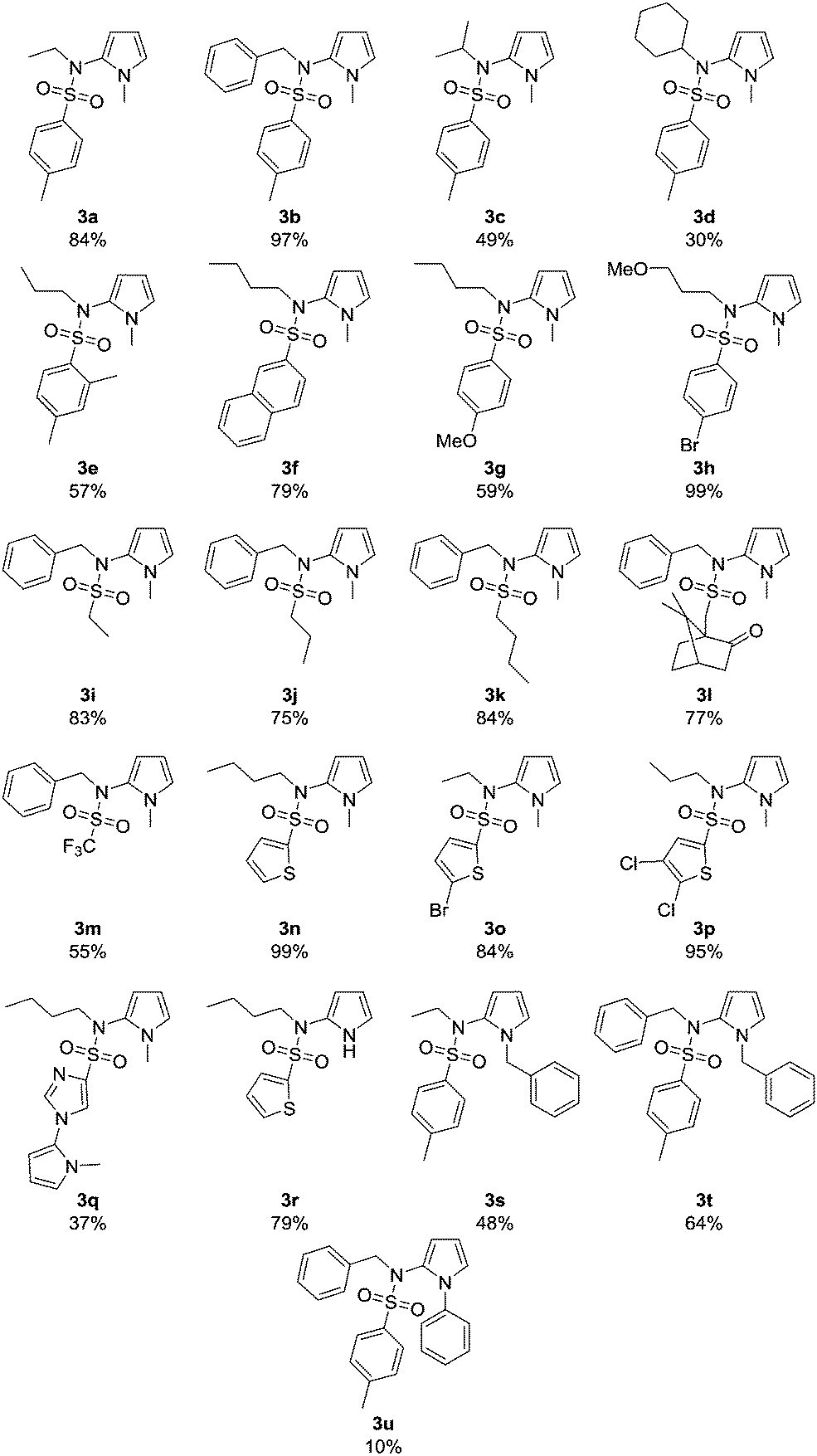
The scope of the reaction was explored using the optimized reaction conditions (Table 1, entry 3): various sulfonamides 1, N-substituted pyrroles 2 (5–20 equiv.), 10 mol% 9-mesityl-10-methylacridinium perchlorate (A), blue light irradiation, oxygen as terminal oxidant, sodium hydroxide (2 equiv.) as base and acetonitrile/water (3:1) as solvent mixture. As depicted in Table 2, all expected products 3a–v were obtained (yield 10–99%). N-Me-pyrrole (2a) reacted with various sulfonamides 1 in moderate to excellent yields of 30–99%. The R1 moiety can be an aromatic group (toluene 3a–3d, m-xylene 3e, naphthalene 3f, 4-methoxy-benzene 3g, and 4-bromo-benzene 3h), an alkyl rest (primary alkyl chains 3i–3k, the bulky 10-camphor 3l, and trifluoromethane 3m) or a heteroarene (thiophene derivatives 3n–3p, and imidazole 3q). The reaction with the imidazole sulfonamide 1q resulted in two C–N bond formations (3q), as the base can deprotonate the imidazole moiety, which reacts with a second molecule 2a. A benzyl-trifluorosulfonamide group was introduced to N-methylpyrrole in compound 3m in 55% yield. The R2 group was varied using different primary (3a, 3e–3h, 3n–3q) and secondary alkyl chains (3c, 3d) and benzyl (3b, 3i–3m). Electron donating or electron withdrawing substituents are generally well tolerated. The bromide and chloride substituents (3h, 3o, 3p) allow further synthetic modifications of the coupling products. Sulfonamides 1r–1t with R2 = phenyl are not converted as their anions are less nucleophilic. Several pyrrole derivatives, such as 2b, N-benzylpyrrole (2c) and 1-phenylpyrrole (2d) led to the products 3r (79%), 3s (48%), 3t (64%), and 3u (10%), respectively. The molecular structures of compounds 3b–3d, 3j, and 3r were confirmed by X-ray single crystal analysis (Fig. 2).
|
|
|---|
|

 | ||
| Fig. 2 Crystal structures of compounds 3b–3d, 3j and 3r. | ||
While a wide variety of N-alkyl sulfonamides can be used in the reaction, the scope of the heterocycle undergoing C–N arylation is limited. The sulfonamidation proceeds selectively with pyrroles; the reaction under identical conditions using furan, thiophene, indole, anisol or dimethoxybenzene does not yield the expected product and starting materials are re-isolated. In a reaction mixture with 2a and furan (1:1), product 3a is formed exclusively from 1a in comparable yield to a reaction in absence of furan. This high specificity of the reaction can be explained by the limited oxidation power of the excited acridinium photocatalysts and the required sufficient stability of the heterocycle and its radical cation under the reaction conditions for a clean conversion with sulfonamide anions as nucleophiles.41 The formation of specific aggregates can also not be excluded.
In 2004 Fukuzumi et al. reported an excited state reduction potential (Ered*) of 1.88 vs. SCE (in PhCN)23 for the charge transfer triplet (CTT) state of 9-mesityl-10-methylacridinium. Verhoeven et al. stated Ered* = 1.45 V vs. SCE (in MeCN) for the locally excited triplet (LET) state.42 The dye has been very well investigated in the last decade revealing a charge transfer singlet (CTS) state with Ered* = 2.08 V vs. SCE (in MeCN) and a locally excited singlet (LES) state with Ered* = 2.18 V vs. SCE (in MeCN).23,42–45 Nicewicz et al. used acridinium dyes to oxidize alkenes24,46–50 and electron-rich arenes27 to their corresponding radical cations and subsequently trapped them with suitable nucleophiles. Our proposed mechanism (Fig. 3) is based on the reported catalytic cycles and mechanistic investigations, and is supported by cyclic voltammetry measurements. Photocatalyst A is excited by blue light irradiation. N-Methylpyrrole (2a) has an oxidation potential of 1.20 V vs. SCE (in MeCN, see ESI†) and can be therefore easily oxidized by A* to 2a˙+. Oxygen regenerates A resulting in the formation of superoxide O2˙−.27,43,45,51 Under the reaction conditions, sulfonamide 1 is partly deprotonated by sodium hydroxide and the resulting anion 1− reacts as nucleophile with the radical cation of 2a. The process was studied in detail by Moeller et al. for the intramolecular reaction between radical cations of alkenes and sulfonamides.19–22 Superoxide O2˙− may abstract a hydrogen from 3˙ yielding the desired product 3.27 Alternatively, oxidation and deprotonation steps may yield the product.
 | ||
| Fig. 3 Proposed catalytic cycle for the visible light-mediated C–H sulfonamidation of N-methylpyrrole (2a). | ||
The excited state of the organic dye 9-mesityl-10-methylacridinium (A) is able to oxidize pyrroles to the corresponding radical cation, which is subsequently attacked by sulfonamide anion nucleophiles. Reoxidation and deprotonation or hydrogen atom transfer from the resulting radical intermediate results in products of an oxidative C–H sulfonamidation in the 2-position of pyrrole. The mild metal free photocatalytic oxidation protocol does not require prefunctionalized starting materials such as aryl halides or arylboronic acids or the use of less stable sulfonyl azides. A plausible reaction mechanism was proposed and is supported by electrochemical investigations. The method may find use in the synthesis of functionalized N-pyrrole sulfonamides, which are interesting structures with potential pharmaceutical activity.
This work was supported by the German Science Foundation (DFG) (GRK 1626). A. M. thanks the Fonds der Chemischen Industrie for a scholarship. We thank Dr Rudolf Vasold for GC-MS measurements and Ms Regina Hoheisel for cyclic voltammetry measurements.
Notes and references
- H. Rojas Cabrera, G. Huelgas, J. M. Hernández Pérez, P. J. Walsh, R. Somanathan and C. Anaya de Parrodi, Tetrahedron: Asymmetry, 2015, 26, 163–172 CrossRef.
- S. R. Dubbaka and P. Vogel, Angew. Chem., Int. Ed., 2005, 44, 7674–7684 CrossRef PubMed.
- A. Ritzén, M. D. Sørensen, K. N. Dack, D. R. Greve, A. Jerre, M. A. Carnerup, K. A. Rytved and J. Bagger-Bahnsen, ACS Med. Chem. Lett., 2016, 7, 641–646 CrossRef PubMed.
- T. A. Stammers, R. Coulombe, J. Rancourt, B. Thavonekham, G. Fazal, S. Goulet, A. Jakalian, D. Wernic, Y. Tsantrizos, M.-A. Poupart, M. Bös, G. McKercher, L. Thauvette, G. Kukolj and P. L. Beaulieu, Bioorg. Med. Chem. Lett., 2013, 23, 2585–2589 CrossRef PubMed.
- E. A. Wydysh, S. M. Medghalchi, A. Vadlamudi and C. A. Townsend, J. Med. Chem., 2009, 52, 3317–3327 CrossRef PubMed.
- F. Weber and G. Sedelmeier, Nachr. Chem., 2013, 61, 528–529 CrossRef.
- F. Weber and G. Sedelmeier, Nachr. Chem., 2014, 62, 997 CrossRef.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed., 1995, 34, 1348–1350 CrossRef.
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef.
- G. Burton, P. Cao, G. Li and R. Rivero, Org. Lett., 2003, 5, 4373–4376 CrossRef PubMed.
- S. Fu, H. Jiang, Y. Deng and W. Zeng, Adv. Synth. Catal., 2011, 353, 2795–2804 CrossRef.
- H. Chen and M. P. Huestis, ChemCatChem, 2015, 7, 743–746 CrossRef.
- B. Zhu, X. Cui, C. Pi, D. Chen and Y. Wu, Adv. Synth. Catal., 2016, 358, 326–332 CrossRef.
- Z. Song and A. P. Antonchick, Org. Biomol. Chem., 2016, 14, 4804–4808 Search PubMed.
- W.-C. C. Lee, Y. Shen, D. A. Gutierrez and J. J. Li, Org. Lett., 2016, 18, 2660–2663 CrossRef PubMed.
- Y.-X. Li, H.-X. Wang, S. Ali, X.-F. Xia and Y.-M. Liang, Chem. Commun., 2012, 48, 2343–2345 RSC.
- B. Prasad, B. Y. Sreenivas, D. Rambabu, G. R. Krishna, C. Malla Reddy, K. L. Kumar and M. Pal, Chem. Commun., 2013, 49, 3970–3972 RSC.
- S. Badigenchala, V. Rajeshkumar and G. Sekar, Org. Biomol. Chem., 2016, 14, 2297–2305 CAS.
- H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130, 13542–13543 CrossRef CAS PubMed.
- H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132, 2839–2844 CrossRef CAS PubMed.
- H.-C. Xu and K. D. Moeller, Org. Lett., 2010, 12, 1720–1723 CrossRef CAS PubMed.
- J. M. Campbell, H.-C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2012, 134, 18338–18344 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- Visible light C–H amidation of heteroarenes: E. Brachet, T. Ghosh, I. Ghosh and B. König, Chem. Sci., 2015, 6, 987–992 RSC.
- Photoredox catalyzed aryl amination with nickel salts and an iridium complex: E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279–283 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC.
- G. S. Basarab, P. J. Hill, A. Rastagar and P. J. H. Webborn, Bioorg. Med. Chem. Lett., 2008, 18, 4716–4722 CrossRef CAS PubMed.
- M. T. Huggins, T. Butler, P. Barber and J. Hunt, Chem. Commun., 2009, 5254–5256 RSC.
- M. M. Ghorab, F. A. Ragab, H. I. Heiba, H. A. Youssef and M. G. El-Gazzar, Bioorg. Med. Chem. Lett., 2010, 20, 6316–6320 CrossRef CAS PubMed.
- M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed.
- R. C. C. Carvalho, W. A. Martins, T. P. Silva, C. R. Kaiser, M. M. Bastos, L. C. S. Pinheiro, A. U. Krettli and N. Boechat, Bioorg. Med. Chem. Lett., 2016, 26, 1881–1884 CrossRef CAS PubMed.
- B. D. Roth, C. J. Blankley, A. W. Chucholowski, E. Ferguson, M. L. Hoefle, D. F. Ortwine, R. S. Newton, C. S. Sekerke, D. R. Sliskovic and M. Wilson, J. Med. Chem., 1991, 34, 357–366 CrossRef CAS PubMed.
- Penta-substituted pyrroles having a sulfonamide-moiety in position 2 can be synthesized via gold-catalysis: Y. Wu, L. Zhu, Y. Yu, X. Luo and X. Huang, J. Org. Chem., 2015, 80, 11407–11416 CrossRef CAS PubMed.
- Penta-substituted pyrroles having a sulfonamide-moiety in position 2 can be synthesized via gold-catalysis: S. K. Pawar, R. L. Sahani and R.-S. Liu, Chem. – Eur. J., 2015, 21, 10843–10850 CrossRef CAS PubMed.
- F. Teplý, Collect. Czech. Chem. Commun., 2011, 76, 859–917 CrossRef.
- A. U. Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050–2054 CrossRef CAS.
- A. U. Meyer, K. Straková, T. Slanina and B. König, Chem. – Eur. J., 2016, 22, 8694–8699 CrossRef CAS PubMed.
- A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
- Indole derivatives are having a suitable oxidation potential, but they are not stable under the basic conditions.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, H. J. van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 127, 16054–16064 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- T. Hering, T. Slanina, A. Hancock, U. Wille and B. König, Chem. Commun., 2015, 51, 6568–6571 RSC.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed.
- P. D. Morse and D. A. Nicewicz, Chem. Sci., 2015, 6, 270–274 RSC.
- N. J. Gesmundo, J.-M. M. Grandjean and D. A. Nicewicz, Org. Lett., 2015, 17, 1316–1319 CrossRef CAS PubMed.
- K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures, synthesis, spectroscopic, and crystallographic data. CCDC 1494732–1494736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06111g |
| ‡ Both authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |