
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding memory effects in Li-ion batteries: evidence of a kinetic origin in TiO2 upon hydrogen annealing†
E.
Ventosa
*a,
T.
Löffler
a,
F.
La Mantia
bc and
W.
Schuhmann
 a
a
aAnalytical Chemistry – Center for Electrochemical Sciences (CES), Ruhr-Universität Bochum, D-44780 Bochum, Germany. E-mail: edgar.ventosa@rub.de
bSemiconductor and Energy Conversion – Center for Electrochemical Sciences (CES), Ruhr-Universität Bochum, Universitätstr. 150, D-44780 Bochum, Germany
cEnergiespeicher- und Energiewandlersysteme, Universität Bremen, Wiener Str. 12, 28359 Bremen, Germany
First published on 22nd August 2016
Abstract
Memory effects in Li-ion battery materials have been explained on the basis of the thermodynamics of many-particles body, however the role of the (de-)intercalation kinetics is not yet clear. We demonstrate that kinetic aspects, specifically Li-ion mobility, are determining the magnitude of the memory effect in TiO2 by studying samples with different levels of oxygen vacancies.
In batteries such as Ni–Cd or Ni–MH the (dis-)charge potential profile is influenced by their history. In other words, these batteries “memorize” the history of (dis-)charge, e.g. depth of charge or discharge in previous cycles, and they behave differently according to that history.1–3 This effect has critical consequences on the performance of a battery as it interferes with the readings of the state of charge (SOC). This issue was believed to be overcome in Li-ion batteries (LIBs) since intercalation materials were reported not to suffer from memory effects, which was seen as one of the advantages of LIBs.4 However, Sasaki et al. revealed the presence of a memory effect in LiFePO4 which is one of the most widely used battery material.5 This phenomenon was also detected in other intercalation materials, i.e. TiO2 and Al-doped Li4Ti5O12.6,7 So far, little is known about the origin and mechanism of the memory effect in intercalation materials. In a first attempt, the memory effect was hypothesized to originate from the particle-by-particle charging process, which derives from the two-phase (Li-rich and Li-poor) equilibrium of LiFePO4. This proposed mechanism was entirely based on the thermodynamics of intercalation materials, and could not properly explain empiric observations such as a dependence of the magnitude of the memory effect on the particle size of TiO2,6 or the appearance of a memory effect upon Al-doping in Li4T5O12.7 Here, we investigate the kinetic origin of the memory effect in LIBs using anatase TiO2 as model material, specifically the influence of the Li-ion mobility in the intercalation material on the memory effect.
The high operating potential of TiO2 (ca. 1.8 V vs. Li/Li+) reduces the cell voltage and consequently the energy density of the resulting battery. However, it offers several advantages. The large voltage gap to Li electroplating enhances the safety, while the absence of a thick SEI increases the cyclability.8,9 The latter also makes TiO2 a good model material due to its relatively simple solid-liquid interface. The slow kinetics of Li-ion (de-)intercalation in TiO2 impede achieving its theoretical specific charge of 336 mA h g−1 at moderate C-rates (>0.1C). Both Li-ion and electron conductivities in TiO2 are relatively low as compared to other intercalation materials. Many strategies have been reported to improve the kinetics of (de-)intercalation in TiO2.9 Introducing oxygen vacancies (TiO2−x) by hydrogen annealing of anatase TiO2 has been demonstrated to be an effective and simple approach.10–13 Specifically, the electron mobility is enhanced with increasing levels of oxygen vacancies while Li-ion mobility decreases, as illustrated in Scheme 1.10 As a result, the kinetics of (de-)intercalation improve with increasing levels of oxygen vacancies until reaching a maximum. After that, the decrease in Li-ion mobility outweighs the increase in electron mobility.10 The changes in (de-)intercalation kinetics upon introduction of oxygen vacancies are used in this work to elucidate the effect of (de-)intercalation kinetics on the memory effect.
 | ||
| Scheme 1 Illustration of the changes in electron and Li-ion mobility in TiO2 upon increasing oxygen vacancies. Adapted with permission from ref. 10. | ||
Commercially available anatase TiO2 (TiO2_pristine) was annealed in H2 at 300 °C for 4 h (TiO2_4 h) and 24 h (TiO2_24 h). X-ray diffraction (XRD) of TiO2_24 h ruled out the appearance of new crystal phase even after annealing for 24 h (see ESI,† Fig. S1a). All three samples possessed high specific surface areas (>200 m2 g−1), namely, 300 m2 g−1, 220 and 210 m2 g−1 for TiO2_pristine, TiO2_4 h and TiO2_24 h, respectively. The darkening in the color of the samples upon prolonged annealing time (Fig. 1) indicates a gradual increase in the level of oxygen vacancies. UV-Vis spectroscopy confirmed the introduction of oxygen vacancies (see ESI,† Fig. S1b).
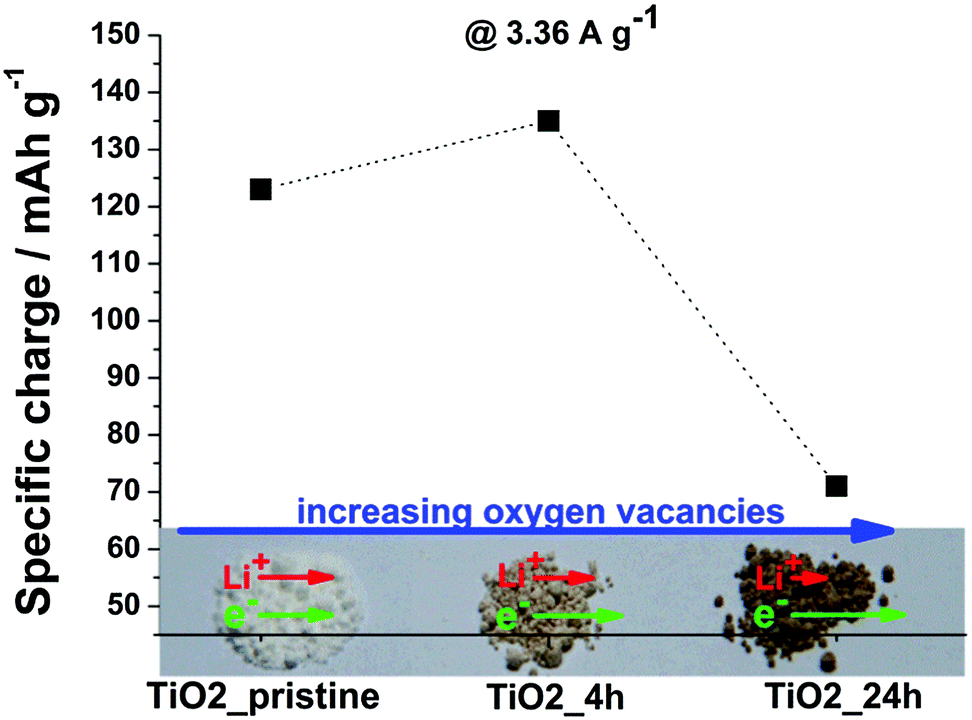 | ||
| Fig. 1 Specific charge at 3.36 A g−1 (theoretical C-rate of 10 C) as well as photograph of TiO2_pristine, TiO2_4 h and TiO2_24 h. | ||
The specific charge delivered at high C-rates (3.36 A g−1 corresponding to a theoretical C-rate of 10 C) is dependent on the kinetics of (de-)intercalation: the faster the kinetics, the higher is the specific charge. Fig. 1 shows that the kinetics in TiO2 slightly improved for the sample annealed for 4 h, but drastically decreased in case of a 24 h treatment. This trend primary derives from the introduction of oxygen vacancies in TiO2 upon H2 annealing, as shown in previous reports.10–12 The specific charge increases from TiO2_pristine to TiO2_4 h due to the increased electric conductivity and it decreases for prolonged annealing time (TiO2_24 h) since the decrease in Li-ion mobility outweighs the increase in electric conductivity.10 The contribution of the changes in specific surface area to the trend is negligible (see ESI† for further discussion).
An electrochemical protocol was used to determine the presence and magnitude of memory effect.5–7 The protocol consisted of applying three subsequent cycles: a normal (de-)intercalation cycle, a memory writing cycle and a memory releasing cycle (Fig. 2a). During the memory writing cycle, the anodic process was interrupted at a certain SOC. In the next memory releasing step, the sample was cycled within the normal potential range, and the memory effect was revealed if occurred. It appeared as a bump in the plateau on Li-ion de-intercalation occurring at a similar SOC at which the memory writing cycle was stopped (Fig. 2a and b). The magnitude of the memory effect was defined as the difference (in mV) between the potential of the maximum of the memory effect bump and the potential at that SOC during the normal cycle (Fig. 2b).
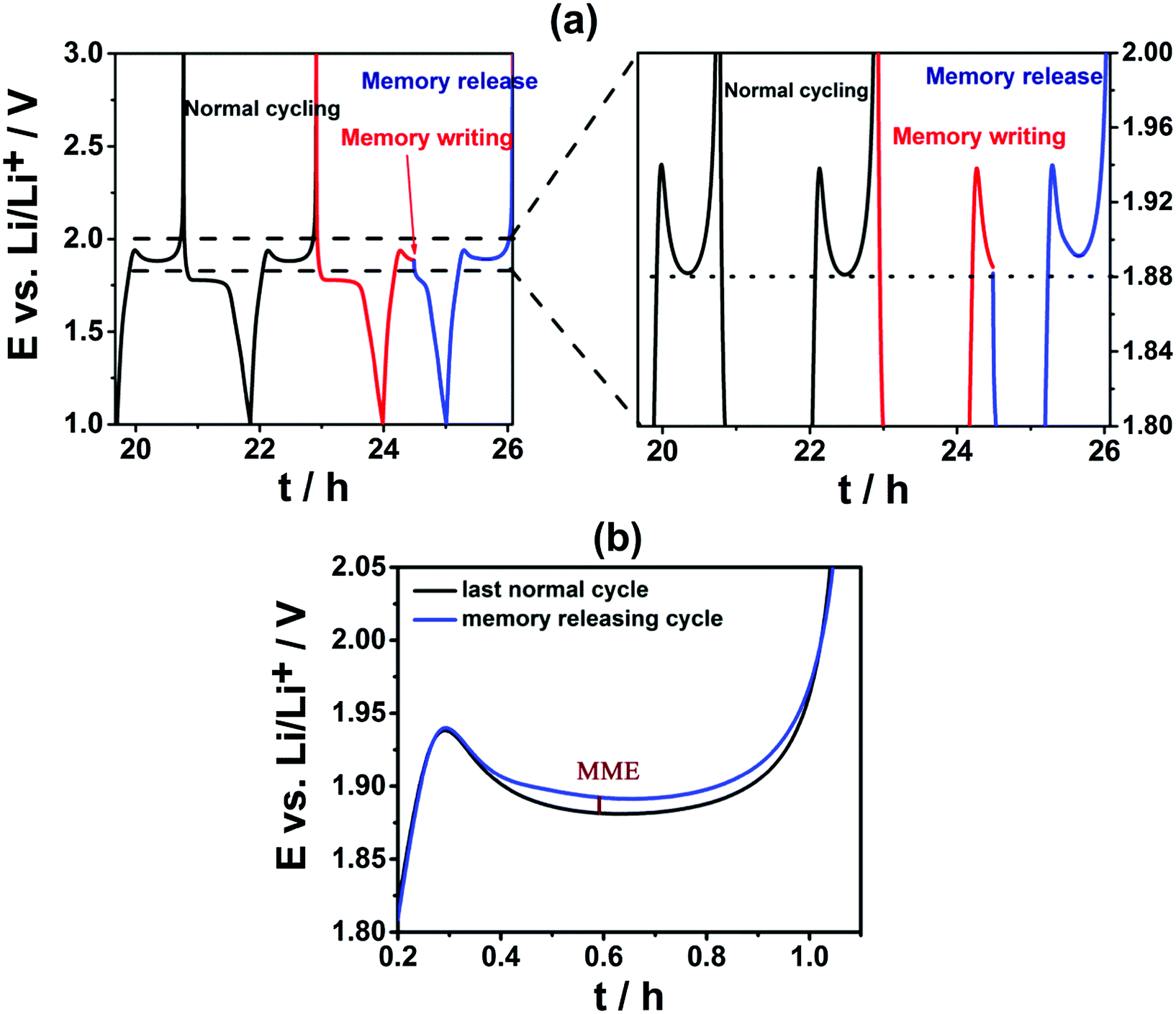 | ||
| Fig. 2 (a) Potential profile of TiO2 during the three subsequent cycles required for the determination of the magnitude of the memory effect (MME). (b) Superimposed potential profiles of a normal cycle and a memory releasing cycle. | ||
The magnitude of the memory effect (MME) is known to increase with decreased specific surface area.6 Due to the changes in specific surface area (300, 220 and 210 m2 g−1 for TiO2_pristine to TiO2_4 h and TiO2_24 h, respectively), a slight increase in MME was expected from TiO2_pristine to TiO2_4 h, while remaining almost constant between TiO2_4 h and TiO2_24 h. However, the MME showed only a small decrease between TiO2_pristine and TiO2_4 h, but it drastically increased for TiO2_24 h (Fig. 3). This trend in the MME completely matched that of the specific charge (Fig. 2) indicating that the MME is strongly correlated with the kinetics of (de-)intercalation process: the faster the kinetics the smaller the memory effect. In this particular case, the sluggish Li-ion mobility in TiO2 after H2 annealing for 24 h was responsible for the significant increase in the MME since the electron conductivity continuously increases upon H2 annealing.10 Another commercial anatase TiO2 with lower specific surface area (95 m2 g−1) was also evaluated. In this case, the influence of the specific surface area was completely ruled out since it did not change upon annealing. The exact same trend in the MME was observed for TiO2 with 95 m2 g−1 confirming the influence of oxygen vacancies on MME. In addition, for the same annealing time, the MME for smaller surface area TiO2 (95 m2 g−1) was always higher than that of its homologous sample of higher surface area (300 m2 g−1) confirming the dependency between specific surface area and memory effect. Since it is known that larger particles lead to slower kinetics,8,9,14,15 the higher memory effect for larger particles can be explained on the basis of their slower kinetics. Also the appearance of the memory effect on Al-doped Li4Ti5O12 (pristine Li4Ti5O12 does not suffer from any memory effect) can be attributed to the slower Li-ion mobility upon doping.
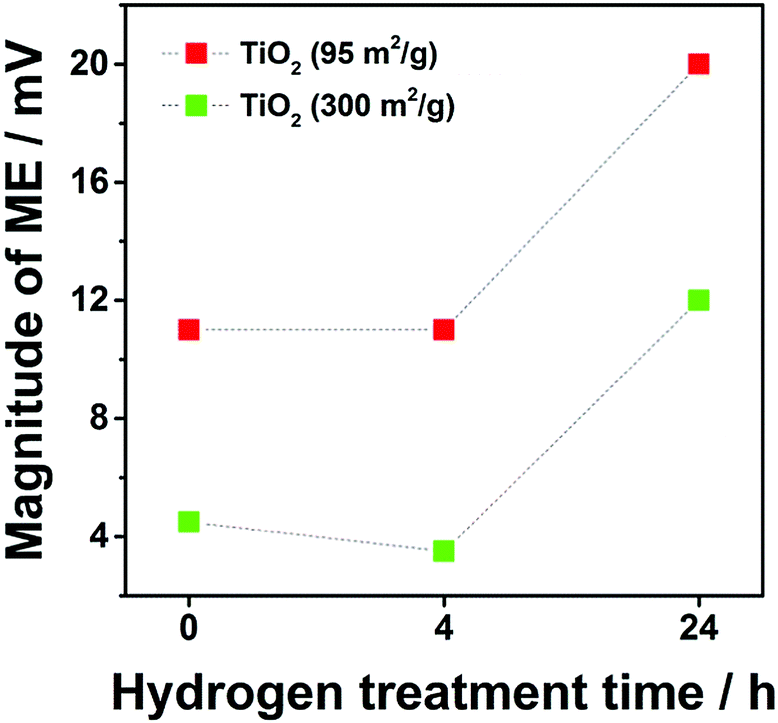 | ||
| Fig. 3 Dependence of the magnitude of the memory effect (MME) on the duration of H2 annealing time (pristine, 4 h and 24 h) for two commercial TiO2 samples with 300 m2 g−1 in green and with 95 m2 g−1 in red. The potential profiles of all samples are shown in Fig. S2 (ESI†). | ||
Regarding the overshoot (small peak in the anodic galvanostatic curves before the plateau, Fig. 2b), the relationship between Li-ion mobility, overshoot and memory effect was found to be highly non-linear and saturate as discussed in ESI† (Fig. S3).
In conclusion, while thermodynamics of materials define whether a Li-ion battery material could suffer from memory effect, kinetic aspects have a great influence on whether the phenomenon is observed or not. In the studied case, the slower kinetics related to hindered Li-ion mobility in TiO2 is causing an increase of the MME. Other reported observations can now be explained on the basis of the kinetics of (de-)intercalation, such as the dependency between particle size and memory effect or the appearance of memory effect upon doping for pristine Li4Ti5O12 and Al-doped Li4Ti5O12.
References
- D. Linden, T. B. Reddy, Handbook of Batteries, McGraw-Hill, New York, 2002, pp. 28–18, ISBN 0-07-135978-8 Search PubMed.
- R. A. Huggins, Solid State Ionics, 2006, 177, 2643 CrossRef CAS.
- Y. Sato, S. Takeuchi and K. Kobayakawa, J. Power Sources, 2001, 93, 20 CrossRef CAS.
- Y. Nishi, J. Power Sources, 2001, 100, 101 CrossRef CAS.
- T. Sasaki, Y. Ukyo and P. Novak, Nat. Mater., 2013, 12, 569 CrossRef CAS PubMed.
- E. Madej, F. La Mantia, W. Schuhmann and E. Ventosa, Adv. Energy Mater., 2014, 4, 1400829 CrossRef.
- D. Li, Y. Sun, X. Liu, R. Peng and H. Zhou, Chem. Sci., 2015, 6, 4066 RSC.
- Z. Chen, I. Belharouak, Y.-K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23, 959 CrossRef CAS.
- M. Fehse and E. Ventosa, ChemPlusChem, 2015, 80, 785 CrossRef CAS.
- J.-Y. Shin, J. H. Joo, D. Samuelis and J. Maier, Chem. Mater., 2012, 24, 543 CrossRef CAS.
- E. Ventosa, W. Xia, S. Klink, F. La Mantia, B. Mei, M. Muhler and W. Schuhmann, Chem. – Eur. J., 2013, 19, 14194 CrossRef CAS PubMed.
- T. Xia, W. Zhang, J. B. Murowchick, G. Liu and X. Chen, Adv. Energy Mater., 2013, 3, 1516 CrossRef CAS.
- E. Ventosa, A. Tymoczko, K. Xie, W. Xia, M. Muhler and W. Schuhmann, ChemSusChem, 2014, 7, 2584 CrossRef CAS PubMed.
- M. Wagemaker, W. J. H. Borghols and F. M. Mulder, J. Am. Chem. Soc., 2007, 129, 4323 CrossRef CAS PubMed.
- C. Jiang, M. Wei, Z. Qi, T. Kudo, I. Honma and H. Zhou, J. Power Sources, 2007, 166, 239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details about the chemicals, the sample preparation, the electrochemical measurements, XRD patterns and UV-Vis spectra. See DOI: 10.1039/c6cc06070f |
| This journal is © The Royal Society of Chemistry 2016 |