
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Genetically encoded fluorophenylalanines enable insights into the recognition of lysine trimethylation by an epigenetic reader†
Yan-Jiun
Lee
a,
M. J.
Schmidt
b,
Jeffery M.
Tharp
a,
Annemarie
Weber
b,
Amber L.
Koenig
c,
Hong
Zheng
d,
Jianmin
Gao
d,
Marcey L.
Waters
c,
Daniel
Summerer
*e and
Wenshe R.
Liu
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 7743, USA. E-mail: wliu@chem.tamu.edu
bDepartment of Chemistry, University of Konstanz, 78457 Konstanz, Germany
cDepartment of Chemistry, Boston College, Boston, MA 02467, USA
dDepartment of Chemistry, University of North Carolina, Chapel Hill, NC 27599, USA
eDepartment of Chemistry and Chemical Biology, Technical University of Dortmund, 44227 Dortmund, Germany. E-mail: Daniel.summerer@tu-dortmund.edu
First published on 27th September 2016
Abstract
Fluorophenylalanines bearing 2–5 fluorine atoms at the phenyl ring have been genetically encoded by amber codon. Replacement of F59, a phenylalanine residue that is directly involved in interactions with trimethylated K9 of histone H3, in the Mpp8 chromodomain recombinantly with fluorophenylalanines significantly impairs the binding to a K9-trimethylated H3 peptide.
Due to the size similarity between hydrogen and fluorine atoms, most fluorinated amino acids closely resemble their canonical counterparts. When provided in nutrients, they are usually mistaken as canonical amino acids by the cellular translation system and integrated into proteins at corresponding amino acid sites, leading to mild to severe cellular toxicities.1 Biochemists have long been exploiting this promiscuity of the cellular translation system to generate fluorinated proteins.2 Owing to its high NMR signal that is sensitive toward surrounding environments, fluorine in proteins provides a unique probe to study protein structure and dynamics.3 Fluorine also has a more hydrophobic nature than hydrogen, which endows fluorinated proteins with unique features such as high resistance to denaturants.4 Although convenient in making fluorinated proteins, this residue-specific noncanonical amino acid (ncAA) mutagenesis approach typically leads to partial replacement of a native amino acid as a fluorinated amino acid at multiple sites in a protein and generates a highly heterogeneous final product that leads to complexity in subsequent studies. Consequently, methods for the synthesis of proteins bearing fluorinated amino acids at user-defined sites are of high interest. A successful strategy at this front is the use of amber suppression that in the context of fluorinated amino acids was pioneered by Furter and later expanded by others.5
We previously reported two rationally designed, polyspecific mutants of Methanosarcina mazei pyrrolysyl-tRNA synthetase (PylRS) that enable the effective aminoacylation of tRNAPyl with a large variety of phenylalanine-derived ncAA for their incorporation into proteins by amber suppression. Of these, mutant N346A/C348A/Y306A/Y384F (PylRS-AAAF) accepted phenylalanine derivatives with large substituents at the para position as substrates,6 whereas N346A/C348A (PylRS-AA) accepted small phenylalanine derivatives,7 including the NMR probe m-trifluoromethyl-phenylalanine.
To get insights into the origin of polyspecificity of PylRS-AA and into its lack of phenylalanine recognition, we determined the crystal structure of the C-terminal catalytic fragment (amino acids 188–454)8 in complex with the ATP analog adenosine-5′-(β,γ-imido)triphosphate (AMPPNP) at a resolution of 1.5 Å (the structure of PylRS-AAAF was also solved, for data collection and refinement statistics, see the ESI†).9 In wild type PylRS (PylRS_wt), N346 of the amino acid binding pocket serves as a gatekeeper residue that is engaged in a variety of direct and water-mediated hydrogen bonds (Fig. 1A). This includes donation of one bond to the Nε-carbonyl group of pyrrolysine adenylate, which is believed to be critical for pyrrolysine recognition.10 C348 forms part of the pocket bottom (Fig. 1A). In the structure of PylRS-AA, the mutation of both residues to alanine results in an inability of hydrogen bonding, combined with an enlargement of the pocket, in particular at position 346 (Fig. 1B). A related observation has been made in the context of a different mutant that accepts O-methyl-tyrosine as a substrate.11
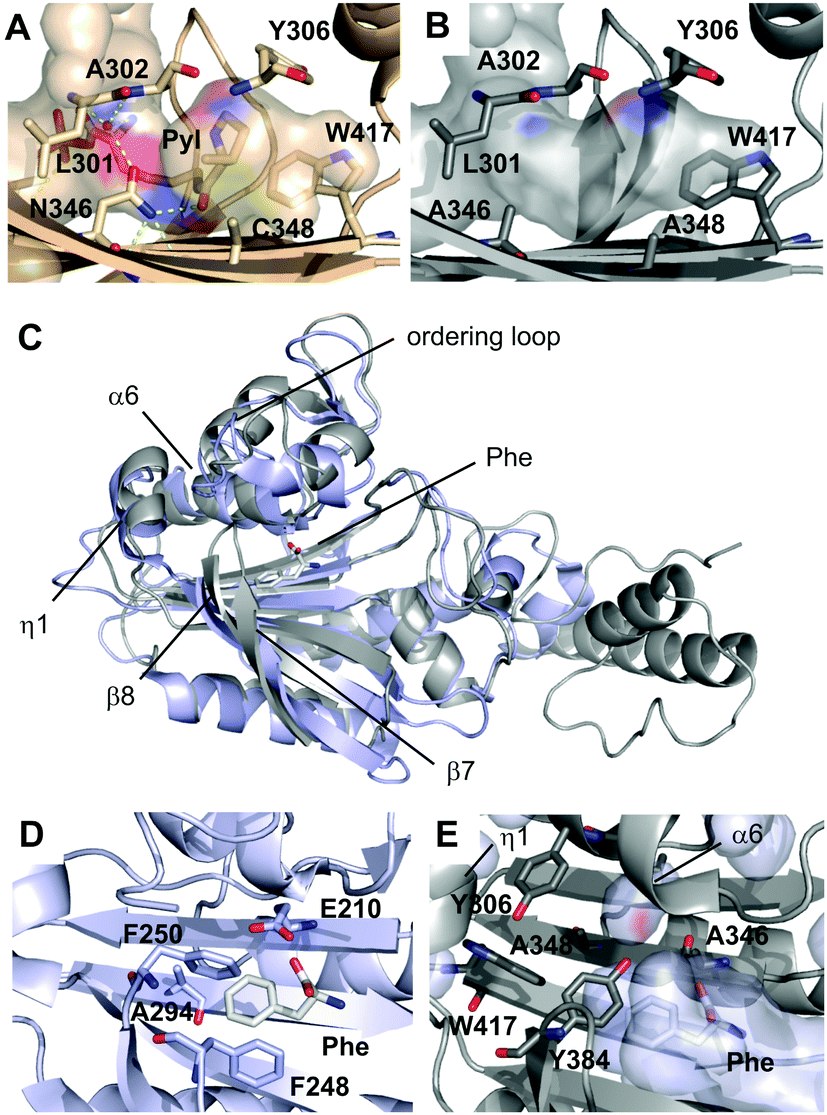 | ||
| Fig. 1 Crystallographic analysis of PylRS-AA. (A) Amino acid binding pocket of PylRS-wt in complex with a pyrrolysyl-adenylate in a previously reported crystal structure (PDB entry: 2Q7H). Hydrogen bonds are shown as dotted yellow lines, water molecule as red sphere. Pocket surface is drawn transparent and colored according to atoms that form the surface. (B) Amino acid binding pocket of PylRS-AA in complex with AMPPNP (PDB entry: 5KIP). Surface color code as in A. (C) Overview of superimposed crystal structures of E. coli PheRS (light blue) in complex with phenylalanine (Phe) and AMP (pdb entry 3PCO) and PylRS_AA (grey) in complex with AMPPNP. The Phe ligand of PheRS is shown as sticks. (D) Amino acid binding pocket of PheRS with pocket-forming residues and Phe ligand shown as sticks. (E) Amino acid binding pocket of PylRS-AA with pocket-forming residues shown as sticks. PheRS structure was superimposed and Phe ligand (white sticks) and PheRS pocket surface (transparent light blue) are shown. | ||
PylRS belongs to the aminoacyl-tRNA-synthetase subclass IIc that also includes PheRS from that PylRS has directly evolved, and both show similarity in their overall fold and in the organization of the core domain (Fig. 1C).9 Our structure of PylRS-AA reveals similarities with PheRS in the front pocket dimension and polarity, despite marked differences in the type and orientation of the involved residues (Fig. 1D and E). However, the overall pocket dimensions of PylRS-AA (in particular in the rear part) are larger, and in superimposed structures, both the Phe ligand and the pocket surface of PheRS can easily be accommodated in the pocket of PylRS-AA (Fig. 1E). Specifically, in PheRS, residues including E210, F248, F250 and A294 form a very compact binding pocket for phenylalanine (Fig. 1D). Consequently, though PheRS tolerates o, m, and p-fluorophenylalanines as substrates, larger derivatives are usually expelled.12 Residues in PylRS-AA that correspond with E210 and A294 of PheRS are A346 and G41913 that bear no or shorter side chains. L305 in PylRS-AA is a homologous site of E174 in PheRS, but its side chain diverts from the pocket (not shown). Finally, F248 of PheRS has no homologous residue in PylRS-AA although Y384 of PylRS-AA partially occupies its space (Fig. 1D and E). Taken together, the arrangement of active site residues of PylRS-AA leads to an enlarged amino acid binding pocket, and the differential availability of hydrophobic contacts for larger, substituted Phe-derivatives versus unsubstituted Phe may account for the observed selectivity of PylRS-AA (Fig. 5, ESI†).
This structural comparison intrigued us to test the recognition of pentafluoro-phenylalanine (F5F, Fig. 2A) by PylRS-AA. As expected, PylRS-AA does recognize F5F. E. coli BL21 cells coding PylRS-AA, tRNAPyl, and sfGFP2TAG (superfolder green fluorescent protein (sfGFP) with an amber mutation at its S2 position) expressed full-length sfGFP when F5F was provided in the GMML medium, albeit with a low level (Fig. S1, ESI†). In order to identify a PylRS mutant that in coordination with tRNAPyl shows an enhanced amber suppression rate in E. coli for more efficient incorporation of F5F into proteins, we constructed a small PylRS-AA-based mutant library by randomizing A348. A348 is spatially close to E174 in PheRS that locks phenylalanine restrictedly at the PheRS active site. By randomizing A348, we deemed that a better mutant with tighter binding of F5F could be identified. Screening all mutants led to the final identification of the mutant with S348 (coined as PylRS-AS) that in coordination with tRNAPyl provided a higher efficiency of amber suppression in E. coli in the presence of F5F (Fig. S2 and S3, ESI†).
 | ||
Fig. 2 The genetic incorporation of fluorophenylalanines. (A) Structures of five fluorophenylalanines. (B) The expression of sfGFP with fluorophenylalanines incorporated at its S2 position. To express full-length sfGFP, E. coli BL21 cells were transformed with two plasmids coding PylRS-AS, tRNAPyl, and sfGFP2TAG and the transformed cells were grown in the GMML medium supplemented with or without a fluorophenylalanine at 3 mM. (C) Deconvoluted ESI-MS spectra of purified full-length sfGFP proteins. Theoretical molecular weights are 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 819, 27801, 27783, 27765, and 27765 Da for sfGFP-F5F, sfGFP-F4F, sfGFP-F3F, sfGFP-F2F, and sfGFP-F2F′, respectively. 819, 27801, 27783, 27765, and 27765 Da for sfGFP-F5F, sfGFP-F4F, sfGFP-F3F, sfGFP-F2F, and sfGFP-F2F′, respectively. | ||
To test the fidelity of PylRS-AS for the genetic incorporation of F5F in response to the amber codon, E. coli BL21 cells coding for PylRS-AS, tRNAPyl and sfGFP2TAG were grown in GMML medium with or without supplementing F5F. Cells grown in the presence of F5F produced full-length sfGFP (sfGFP-F5F) with an expression level of 10 mg L−1, markedly contrasting to a negligible expression of full-length sfGFP in the absence of F5F (Fig. 2B). This demonstrated that PylRS-AS accepts F5F as substrate but discriminates against canonical amino acids including Phe. Electrospray ionization mass spectrometry (ESI-MS) analysis of the purified sfGFP-F5F displayed a molecular weight of 27817 Da that agreed well with the theoretical mass of 27819 Da. The single dominant ESI-MS peak also indicated that F5F was not recognized by E. coli PheRS, which would lead to replacement of 12 Phe residues in sfGFP during translation. Therefore the genetic encoding of F5F by amber codon is orthogonal to the endogenous translation system.
We next tested the ability of PylRS-AS for the acceptance of other fluorophenylalanines including 2,3,4,5-tetrafluorophenylalaine (F4F),14 3,4,5-trifluorophenylalanine (F3F), 3,5-difluorophenylalanine (F2F), and 3,4-difluorophenylalanine (F2F′). When these fluorophenylalanines were present in the growth medium, E. coli BL21 cells coding for PylRS-AS, tRNAPyl and sfGFP2TAG expressed full-length sfGFP (Fig. 2B). Expression levels under these conditions were similar to the condition with F5F. Molecular weights of purified full-length sfGFP proteins expressed in presence of F4F, F3F, and F2F (sfGFP-F4F, sfGFP-F3F, and sfGFP-F2F, respectively) determined by ESI-MS agreed well with theoretical molecular weights of these proteins (Fig. 2C and Table 1). All three proteins exhibited a single dominant ESI-MS peak, establishing the orthogonality of genetic ncAA incorporation in respect to the endogenous translation system. However, the full-length sfGFP with F2F′ incorporated (sfGFP-F2F′) displayed multiple peaks in its ESI-MS spectrum. The smallest peak at 27765 Da matched the theoretical mass of 27763 Da. However, other peaks were all about multiples of 36 Da addition to the theoretical mass, clearly indicating that F2F′ displaced regular phenylalanine residues in sfGFP. This result demonstrated that the genetic encoding of F2F′ by amber codon is not orthogonal to the endogenous translation system, although PylRS-AS does recognize it as a substrate.
| Theoretical mass (Da) | Detected mass (Da) | |
|---|---|---|
| sfGFP-F5F | 27819 |
27817 |
| sfGFP-F4F | 27801 |
27799 |
| sfGFP-F3F | 27783 |
27783 |
| sfGFP-F2F | 27765 |
27763 |
| sfGFP-F2F′ | 27765 |
27763, 27800, 27835, 27871, 27907 |
In addition to being used as a NMR probe and to improve protein folding, genetically encoded fluorophenylalanines in proteins could potentially be for the investigation of cation–pi interactions such as in the recognition of lysine methylation in histones by epigenetic readers. Being part of epigenetic regulation of chromatin function, histone lysine methylation induces interactions with effector proteins and subsequently regulates DNA replication, repair, and transcription.15 The recognition of methylated lysine typically involves an aromatic cage that has been found in the chromodomain (Fig. 3A), the PHD finger, and the Tudor domain, and appears to be mediated by cation–pi interactions between the methylammonium moiety and aromatic residues in the cage.16 The cation–pi interaction is predominantly electrostatic, occuring between a cation and the quadrupole moment of an aromatic π system (Fig. 3B).17 As the quadrupole moment places partial negative charge above each face of the aromatic ring, favorable interactions with a cation occur perpendicular to the aromatic plane within a typical van der Waals distance. Although a number of theoretical and experimental studies have been carried out to address the importance of the cation–pi interaction in the recognition of lysine methylation,18 it is not clear to what degree the cation–pi interaction contributes to the recognition specificity. A particularly interesting target protein to address this question is the Mpp8 chromodomain (Mpp8C). Mpp8 is a heterochromatin component that specifically recognizes and binds trimethylated K9 of histone H3 and promotes recruitment of proteins that mediate epigenetic repression.19 In Mpp8C, F59 is part of the aromatic cage that directly binds to trimethylated K9 of H3. Replacing this residue with fluorophenylalanines (in particular with F5F that has a strongly reduced partial negative charge above each face of the aromatic side chain) is expected to significantly reduce the binding of Mpp8C to trimethylated K9 of H3 in the case that the cation–pi interaction plays a dominant role. Otherwise, binding would not be strongly affected or might increase due to the more hydrophobic nature of fluorophenylalanines than phenylalanine.
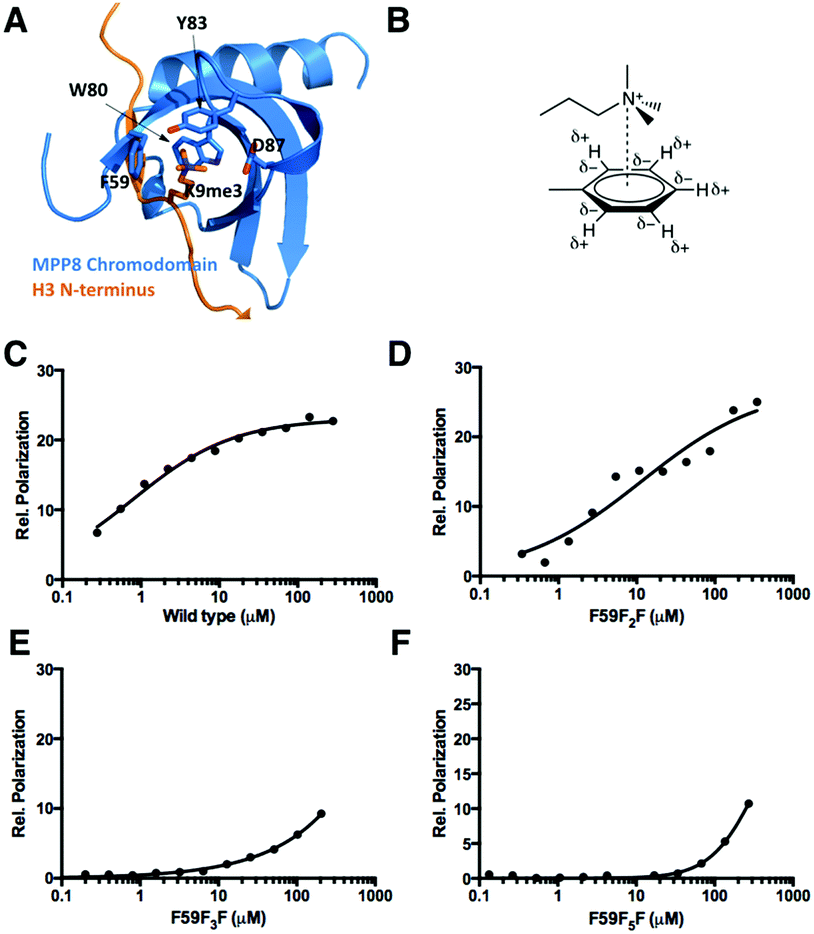 | ||
| Fig. 3 (A) The structure of the MPP8 chromodomain (MPP8C) complexed with the H3(1-15)K9me3 peptide (PDB: 3R93). (B) The cation–quadrupole interaction. (C–F) Fluorescent polarization based binding assays of FAM-H3(1-15)K9me3 interactions with wild type MPP8C, MPP8C-F59F2F, MPP8C-F59F3F, and MPP8C-F5F. Data were fit to the equation: P = Pf + (Pb − Pf) × [protein]/(Kd + [protein]) where Pf and Pb are anisotropies of free and bond ligands. | ||
Using our currently developed approach, Mpp8C with F59 replaced by the three derivatives F5F, F3F, and F2F were expressed. The incorporation of F5F in Mpp8C was independently confirmed with the detection of three 19F NMR singals in the finally purified protein (Fig. S6, ESI†). Together with wild type Mpp8C, interactions of these proteins with a fluorescein-conjugated N-terminal histone H3 peptide with trimethylation at the K9 position (FAM-H3(1-15)K9me3) were studied using fluorescent polarization changes. As shown in Fig. 3C and Table 2, wild type Mpp8C interacts with FAM-H3(1-15)K9me3 strongly, with a determined Kd value around 0.8 μM that agrees with previously reported values.20 This binding was decreased 15-fold when F59 was replaced with F2F and continued to drop when F59 was replaced with F3F and F5F (Fig. 3D–F and Table 2). Due to the low binding of FAM-H3(1-15)K9me3 to both F59F3F and F59F5F mutants of Mpp8C, no sufficient data could be collected to determine accurate Kd values between these two proteins and FAM-H3(1-15)K9me3. This continuous decrease of binding of Mpp8C to FAM-H3(1-15)K9me3 when a growing number of fluorine substituents are added to F59 strongly suggests that the cation–pi interaction plays a dominant role in the binding of trimethylated K9 of H3 to Mpp8C. Though hydrophobic interactions may contribute to the binding, they appear to be not significant, since adding hydrophobicity to F59 does not improve binding.
| K d (μM) | |
|---|---|
| Wild type MPP8C | 0.8 ± 0.1 |
| MPP8C-F59F2F | 12 ± 5 |
| MPP8C-F59F3F | >100 |
| MPP8C-F59F5F | >200 |
In summary, a method for the genetic incorporation of fluorophenylalanines with fluorine substituents at the side chain phenyl ring ranging from 2 to 5 has been developed. This was based on a polyspecific PylRS mutant, its crystal structural analysis, and its further reengineering. The engineered PylRS mutants display recognition of fluorophenylalanines and discriminate against canonical amino acids including phenylalanine, assuring their specific incorporation in response to the amber codon. Using this method, we synthesized Mpp8C, a chromodomain with fluorophenylalanines replacing the critical active site residue F59 that directly interacts with trimethylated K9 of H3 for its binding to Mpp8C. We showed that replacing F59 with fluorophenylalanines significantly weakens the binding of Mpp8C to trimethylated K9 of H3. This result strongly supports a critical involvement of the cation–pi interaction in the recognition of lysine trimethylation by a chromodomain.
This work was supported by National Institute of Health (grants CA161158 to WSL and GM102735 to JG), National Science Foundation (grant CHE-1148684 to WSL, CHE-1112188 to JG, and CHE-1306977 to MLW, and DGE-1144081 to ALK), Welch Foundation (grant A-1715 to WSL), and the Deutsche Forschungsgemeinschaft (SU 726/6-1 in SPP1623).
Notes and references
- (a) R. L. Munier, C. R. Hebd. Seances Acad. Sci., Ser. D, 1959, 248, 1870 CAS; (b) M. J. Pine, Antimicrob. Agents Chemother., 1978, 13, 676 CrossRef CAS PubMed.
- (a) A. Yoshida, Biochim. Biophys. Acta, 1960, 41, 98 CrossRef CAS PubMed; (b) H. R. Arnstein and M. H. Richmond, Biochem. J., 1964, 91, 340 CrossRef CAS PubMed; (c) T. F. Dunn and F. R. Leach, J. Biol. Chem., 1967, 242, 2693 CAS.
- (a) C. Lian, H. Le, B. Montez, J. Patterson, S. Harrell, D. Laws, I. Matsumura, J. Pearson and E. Oldfield, Biochemistry, 1994, 33, 5238 CrossRef CAS PubMed; (b) J. L. Kitevski-Leblanc, F. Evanics and R. Scott Prosser, J. Biomol. NMR, 2010, 47, 113 CrossRef CAS PubMed; (c) J. L. Kitevski-Leblanc, J. Hoang, W. Thach, S. T. Larda and R. S. Prosser, Biochemistry, 2013, 52, 5780 CrossRef CAS PubMed; (d) N. K. Mishra, A. K. Urick, S. W. Ember, E. Schonbrunn and W. C. Pomerantz, ACS Chem. Biol., 2014, 9, 2755 CrossRef CAS PubMed; (e) Y. Suzuki, J. R. Brender, M. T. Soper, J. Krishnamoorthy, Y. Zhou, B. T. Ruotolo, N. A. Kotov, A. Ramamoorthy and E. N. Marsh, Biochemistry, 2013, 52, 1903 CrossRef CAS PubMed.
- (a) C. Yuvienco, H. T. More, J. S. Haghpanah, R. S. Tu and J. K. Montclare, Biomacromolecules, 2012, 13, 2273 CrossRef CAS PubMed; (b) W. L. Hsu, T. C. Shih and J. C. Horng, Biopolymers, 2015, 103, 627 CrossRef CAS PubMed; (c) Y. Tang, G. Ghirlanda, N. Vaidehi, J. Kua, D. T. Mainz, I. W. Goddard, W. F. DeGrado and D. A. Tirrell, Biochemistry, 2001, 40, 2790 CrossRef CAS PubMed; (d) Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado and D. A. Tirrell, Angew. Chem., Int. Ed. Engl., 2001, 40, 1494 CrossRef CAS; (e) A. Niemz and D. A. Tirrell, J. Am. Chem. Soc., 2001, 123, 7407 CrossRef CAS PubMed; (f) T. H. Yoo, A. J. Link and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13887 CrossRef CAS PubMed.
- (a) R. Furter, Protein Sci., 1998, 7, 419 CrossRef CAS PubMed; (b) E. C. Minnihan, D. D. Young, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2011, 133, 15942 CrossRef CAS PubMed; (c) B. J. Wilkins, S. Marionni, D. D. Young, J. Liu, Y. Wang, M. L. Di Salvo, A. Deiters and T. A. Cropp, Biochemistry, 2010, 49, 1557 CrossRef CAS PubMed; (d) M. R. Seyedsayamdost, C. S. Yee and J. Stubbe, Nat. Protoc., 2007, 2, 1225 CrossRef CAS PubMed; (e) T. He, A. Gershenson, S. J. Eyles, Y. J. Lee, W. R. Liu, J. Wang, J. Gao and M. F. Roberts, J. Biol. Chem., 2015, 290, 19334 CrossRef CAS PubMed.
- Y. Kurra, K. A. Odoi, Y. J. Lee, Y. Yang, T. Lu, S. E. Wheeler, J. Torres-Kolbus, A. Deiters and W. R. Liu, Bioconjugate Chem., 2014, 25, 1730 CrossRef CAS PubMed.
- (a) Y. S. Wang, X. Fang, A. L. Wallace, B. Wu and W. R. Liu, J. Am. Chem. Soc., 2012, 134, 2950 CrossRef CAS PubMed; (b) Y. S. Wang, X. Fang, H. Y. Chen, B. Wu, Z. U. Wang, C. Hilty and W. R. Liu, ACS Chem. Biol., 2013, 8, 405 CrossRef CAS PubMed; (c) J. M. Tharp, Y. S. Wang, Y. J. Lee, Y. Yang and W. R. Liu, ACS Chem. Biol., 2014, 9, 884 CrossRef CAS PubMed; (d) A. Tuley, Y. J. Lee, B. Wu, Z. U. Wang and W. R. Liu, Chem. Commun., 2014, 50, 7424 RSC; (e) A. Tuley, Y. S. Wang, X. Fang, Y. Kurra, Y. H. Rezenom and W. R. Liu, Chem. Commun., 2014, 50, 2673 RSC.
- M. J. Schmidt, A. Weber, M. Pott, W. Welte and D. Summerer, ChemBioChem, 2014, 15, 1755 CrossRef CAS PubMed.
- I. Mermershtain, I. Finarov, L. Klipcan, N. Kessler, H. Rozenberg and M. G. Safro, Protein Sci., 2011, 20, 160 CrossRef CAS PubMed.
- J. M. Kavran, S. Gundllapalli, P. O'Donoghue, M. Englert, D. Soll and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11268 CrossRef CAS PubMed.
- J. K. Takimoto, N. Dellas, J. P. Noel and L. Wang, ACS Chem. Biol., 2011, 6, 733 CrossRef CAS PubMed.
- (a) P. Wang, N. Vaidehi, D. A. Tirrell and W. A. Goddard, 3rd, J. Am. Chem. Soc., 2002, 124, 14442 CrossRef CAS PubMed; (b) M. Ibba, P. Kast and H. Hennecke, Biochemistry, 1994, 33, 7107 CrossRef CAS PubMed.
- P. Kast and H. Hennecke, J. Mol. Biol., 1991, 222, 99 CrossRef CAS PubMed.
- (a) L. Qin, C. Sheridan and J. Gao, Org. Lett., 2012, 14, 528 CrossRef CAS PubMed; (b) C. J. Pace, H. Zheng, R. Mylvaganam, D. Kim and J. Gao, Angew. Chem., Int. Ed. Engl., 2012, 51, 103 CrossRef CAS PubMed.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, Nat. Struct. Mol. Biol., 2007, 14, 1025 CAS.
- (a) S. A. Jacobs and S. Khorasanizadeh, Science, 2002, 295, 2080 CrossRef CAS PubMed; (b) J. Mellor, Cell, 2006, 126, 22 CrossRef CAS PubMed; (c) P. R. Nielsen, D. Nietlispach, H. R. Mott, J. Callaghan, A. Bannister, T. Kouzarides, A. G. Murzin, N. V. Murzina and E. D. Laue, Nature, 2002, 416, 103 CrossRef CAS PubMed.
- (a) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed; (b) J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459 CrossRef CAS PubMed; (c) S. Mecozzi, A. P. West, Jr. and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566 CrossRef CAS PubMed.
- (a) R. M. Hughes, K. R. Wiggins, S. Khorasanizadeh and M. L. Waters, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11184 CrossRef CAS PubMed; (b) C. A. Hunter, C. M. Low, C. Rotger, J. G. Vinter and C. Zonta, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4873 CrossRef CAS PubMed; (c) Y. Mo, G. Subramanian, J. Gao and D. M. Ferguson, J. Am. Chem. Soc., 2002, 124, 4832 CrossRef CAS PubMed; (d) C. Ruan and M. T. Rodgers, J. Am. Chem. Soc., 2004, 126, 14600 CrossRef CAS PubMed; (e) F. Costanzo, R. G. Della Valle and V. Barone, J. Phys. Chem. B, 2005, 109, 23016 CrossRef CAS PubMed; (f) A. S. Reddy and G. N. Sastry, J. Phys. Chem. A, 2005, 109, 8893 CrossRef CAS PubMed; (g) Y. Xu, J. Shen, W. Zhu, X. Luo, K. Chen and H. Jiang, J. Phys. Chem. B, 2005, 109, 5945 CrossRef CAS PubMed; (h) N. Yang, W. Wang, Y. Wang, M. Wang, Q. Zhao, Z. Rao, B. Zhu and R. M. Xu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17954 CrossRef CAS PubMed.
- (a) K. Kokura, L. Sun, M. T. Bedford and J. Fang, EMBO J., 2010, 29, 3673 CrossRef CAS PubMed; (b) I. A. Tchasovnikarova, R. T. Timms, N. J. Matheson, K. Wals, R. Antrobus, B. Gottgens, G. Dougan, M. A. Dawson and P. J. Lehner, Science, 2015, 348, 1481 CrossRef CAS PubMed.
- J. Li, Z. Li, J. Ruan, C. Xu, Y. Tong, P. W. Pan, W. Tempel, L. Crombet, J. Min and J. Zang, PLoS One, 2011, 6, e25104 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc05959g |
| This journal is © The Royal Society of Chemistry 2016 |