
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chiroptical molecular sensor for ferrocene†
Marco
Agnes‡
a,
Andrea
Nitti
a,
Douglas A.
Vander Griend
b,
Daniele
Dondi
a,
Daniele
Merli
a and
Dario
Pasini
 *ac
*ac
aDepartment of Chemistry, University of Pavia, Viale Taramelli, 10, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it; Web: http://www.unipv.it/labt
bDepartment of Chemistry & Biochemistry, Calvin College, Grand Rapids, MI 49546-4403, USA
cINSTM Research Unit, University of Pavia, Italy
First published on 10th August 2016
Abstract
A homochiral, square-shaped, D2 symmetrical nanosized metal-linked macrocycle is able to form stable complexes with ferrocene in polar solvents, with detection achieved by means of multiple outputs (optical/chiroptical spectroscopies and cyclic voltammetry). Selective sensing using chiroptical spectroscopy in the presence of interfering analytes is demonstrated.
Chiroptical sensors use their molecular chirality as a tool to detect the binding of a target analyte.1 The most frequent chiroptical sensing mechanisms are either the interaction between a non-racemic chiral substrate and a chromophoric probe, which is silent with respect to circular dichroism (CD) spectroscopy, or the interaction between an achiral analyte and a chromophoric CD-active probe. The appearance or the modulation of the CD signal, respectively, represents the chiroptical readout.2 CD spectroscopy can offer better levels of detection compared with optical spectroscopies and electrochemistry-based methods, and it is frequently used in biosensing, where high sensitivities are required.3
Atropoisomerically-chiral compounds are particularly suitable for application in chiroptical sensing, since the expression of chirality is embedded into a chromophore, resulting in peculiar CD activity.2 In this context, 1,1′-binaphthyl-2,2′-diol (BINOL)-based probes have been previously reported to exhibit a “spring-like” behaviour, with an intense CD signal modulation upon subtle changes in their conformation upon binding.2h,i Such probes have also been incorporated into rigid macrocycles, successfully functioning for the chiroptical detection of species as diverse as anions, cations or C60, either in water or in organic solvents.2f–h,4
Fujita, Stang et al. pioneered the synthetic and supramolecular chemistry of metal-linked macrocycles,5 and in selected cases demonstrated the possibility of recognizing in their cavity electron-rich aromatic compounds with high specificity.6 The extension to chiral metal-linked macrocycles, however, has not been fully addressed.7 In this work, we report the synthesis of conformationally locked metal-linked homochiral macrocycles, and we demonstrate their ability to function as chiroptical sensors towards suitable electron-rich aromatic molecules such as ferrocene. The compounds are assembled using a stable Pd2+–ethylenediamine complex as the angular node, so that pyridine moieties, which replace the two labile cis-ligands of the Pd2+ metal centers, could form metal-linked cyclic species instead of linear coordination polymers.
The m-pyridyl substituted BINOL-based synthon (R)-4, in which different conformations are present as a consequence of the rotation around the aryl–aryl bonds joining the naphthalene and pyridyl fragments, and the p-pyridyl substituted BINOL-based synthon (R)-5 (Fig. 1) were investigated. The treatment of both difunctional ligands with cis-(ethylenediamine)-Pd2+ nitrate salt, under thermodynamically controlled conditions, afforded homochiral metal-containing macrocyclic complexes (RR)-6 and (RR)-7 in high yields, after counterion exchange of the nitrate salts with hexafluorophosphate anions. The optimized molecular structures of the compounds are also shown in Fig. 1.
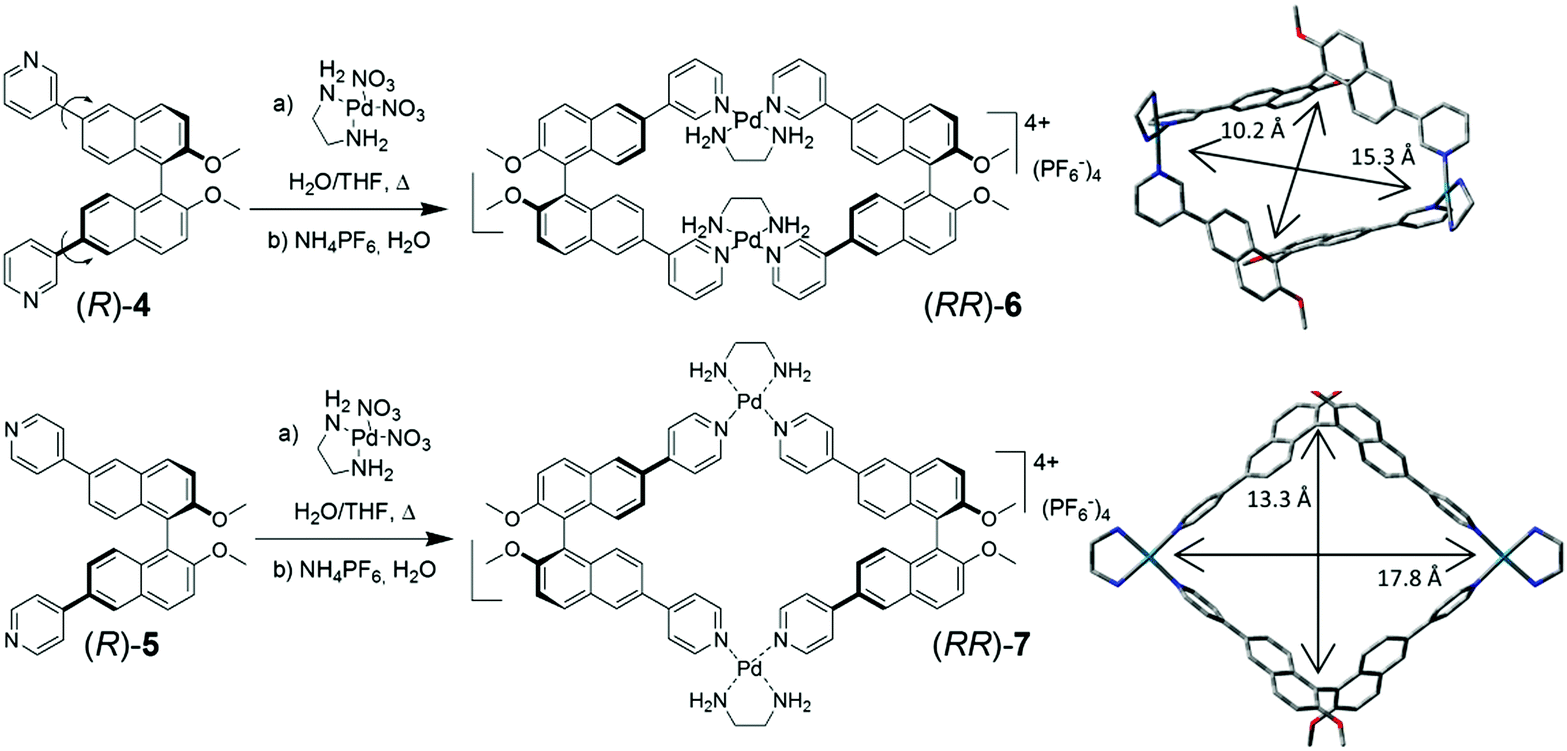 | ||
| Fig. 1 Synthesis of metal-linked macrocycles (RR)-6 and (RR)-7, along with their optimized structures, obtained by molecular modelling. Reported distances refer to the maximum amplitudes of the internal cavities. | ||
The structures of compounds (RR)-6 and (RR)-7 were fully confirmed and characterized by 1H, 13C and 2D NMR spectroscopies (for full experimental details, see the ESI†). The 1H-NMR spectra of the metal-linked macrocycles showed the same sets of signals of the bidentate ligands, consistently with a change in overall symmetry of the molecules from C2 for the ligands to D2 for the macrocycles. The coordinating Pd2+ nuclei clearly enhance the electron-withdrawing character of the pyridyl rings, shifting their proton resonances to higher frequencies (Fig. S1 and S2, ESI†).
MS-ESI spectrometry of both (RR)-6 and (RR)-7 confirmed that for the peak at 1704 m/z, corresponding to the molecular cation [M–PF6]+, the experimental and calculated isotopic patterns were essentially superimposable (Fig. S3, ESI†). This observation rules out the possibility of a doubly charged species (which would result in peak intervals of 0.5 Da), and therefore unequivocally establishes the structure of the complexes as [2 + 2] adducts (composed of two dipyridyl ligands and two metal nodes, as those shown in Fig. 1). Such homochiral low dimensionality was not reported before in similar systems.§
The molecular modelling of (RR)-6 and (RR)-7 highlighted the different dimensions of their internal cavities (Fig. 1).¶ In (RR)-6, the metal corners maintain a perfect square-planar geometry and lock the conformationally-mobile pyridyl moieties on the naphthyl subunits of each ligand facing internally towards each other, with a dihedral angle between the two naphthyl rings of 101°. The near perfect D2 overall symmetry gives rise to a rectangular cavity, with a distance between the BINOL units (measured between the mean of the two naphthalene–naphthalene bonds) of 10.3 Å, defining the narrower aperture of the cavity, while the distance between the two Pd2+ atoms is 15.3 Å. Compound (RR)-7, instead, showed a more square-like geometry in which the naphthalene–naphthalene bonds are 13.3 Å far from each other and the distance between the two Pd2+ atoms is 17.8 Å. The four π-conjugated panels defining the metal-linked macrocycles have, especially in the case of the p-substituted (RR)-7, a “push–pull” character, owing to the metal-coordinated “pull” pyridyl, linked with π-conjugation to the methoxy “push” substituents on the naphthyl rings. The D2 overall symmetry present in both metal-linked macrocycles, however, cancels out the local dipoles generated, giving a “no residual” molecular dipole in both macrocycles.
Although the cavity of (RR)-6 would just be the right fit for aromatic compounds, electron-rich compounds, such as 1,4-dialkoxybenzenes, did not show any binding affinity with either (RR)-6 or, not surprisingly, (RR)-7. Pleasingly, however, ferrocene showed a distinct interaction with the square-shaped macrocycle (RR)-7 in MeCN, which was initially detected using UV/Vis and CD spectroscopies. Representative titrations are shown in Fig. 2. In the UV/Vis titration, a weak but distinctive decrease of the UV/Vis shoulder band at 350 nm, upon titration of a solution of (RR)-7 at constant concentration with increasing amounts of ferrocene, could be clearly observed. Upon complexation, the band at 350 nm also shifted very weakly towards lower energy, indicating that the interactions responsible for the binding did not rely on full, potent π–π stacking interactions, with related charge transfer, between the host and the guest. The binding isotherm (see the inset, Fig. 2 (top)) showed saturation occurring well above 1 equivalent of guest added; furthermore, the absence of an isosbestic point, and the impossibility of fitting the data with the nonlinear equation for the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding pointed strongly towards a situation where multiple equilibria (e.g. the concurrent presence of a 1:2 macrocycle:ferrocene complex) are in place.
:1 binding pointed strongly towards a situation where multiple equilibria (e.g. the concurrent presence of a 1:2 macrocycle:ferrocene complex) are in place.
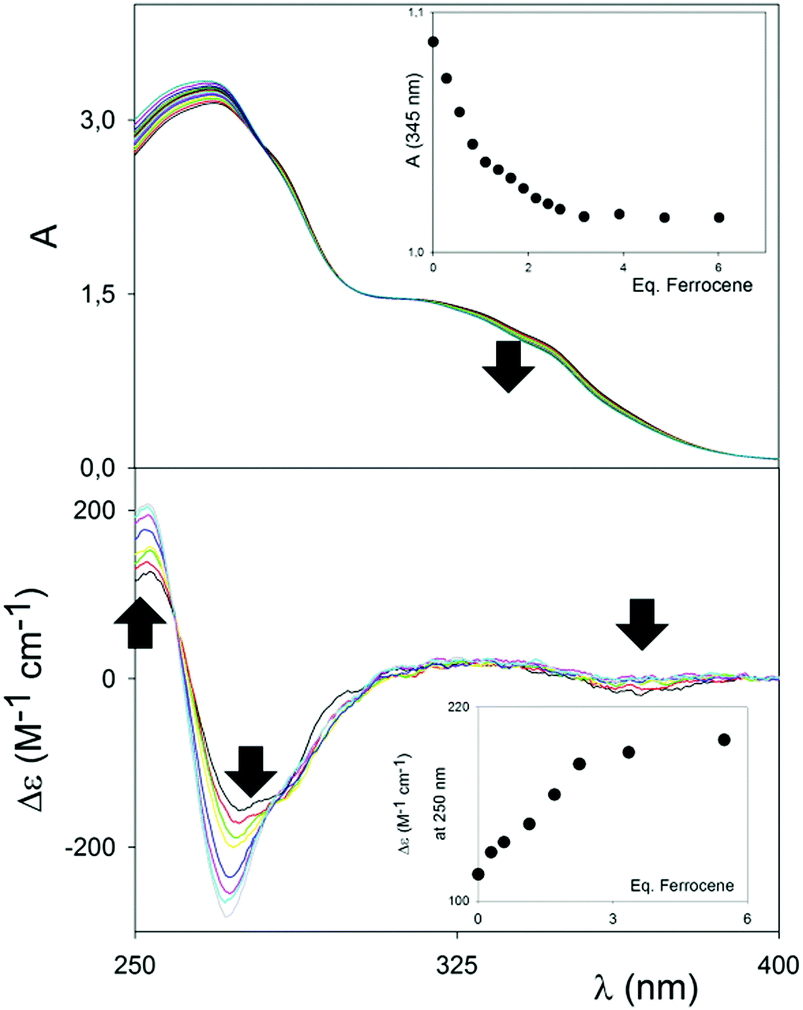 | ||
| Fig. 2 Top: UV titration of metal-linked macrocycle (RR)-7 (2.5 × 10−5 M) with ferrocene in MeCN at 25 °C. Inset: Titration profile vs. ferrocene added equivalents at 345 nm. Bottom: CD titration of macrocycle (RR)-7 (1 × 10−5 M) with ferrocene in MeCN at 25 °C. Inset: Titration profile vs. ferrocene added equivalents at 250 nm. | ||
Variations monitored by CD spectroscopy were found to be much more intense (Fig. 2, bottom).
The exciton couplet band attributable to the π-extended binaphthyl units, centered at 270 nm, corresponding to the λmax of the UV/Vis spectrum, shows a large modulation upon binding, which has to be related to a variation of the dihedral angle between the naphthyl planes of the binaphthyl units, and therefore to a conformational rearrangement of the macrocycle upon binding to the guest. Although the dihedral angle variation and conformational rearrangement are marginal (see modelling in Fig. 3), the ample change in the exciton couple once again reinforces the “spring-like” concept for such axially-chiral probes and it forecasts great utility in sensing. A weak, but detectable increase in the CD band appeared corresponding to the UV/Vis band of the macrocycle/ferrocene complex at ca. 350 nm, which indicates an overall chirality of the complex as a whole. Control titrations with compound (RR)-6 were carried out under identical conditions, and revealed no changes using both UV/Vis and CD spectroscopy (Fig. S4, ESI†).
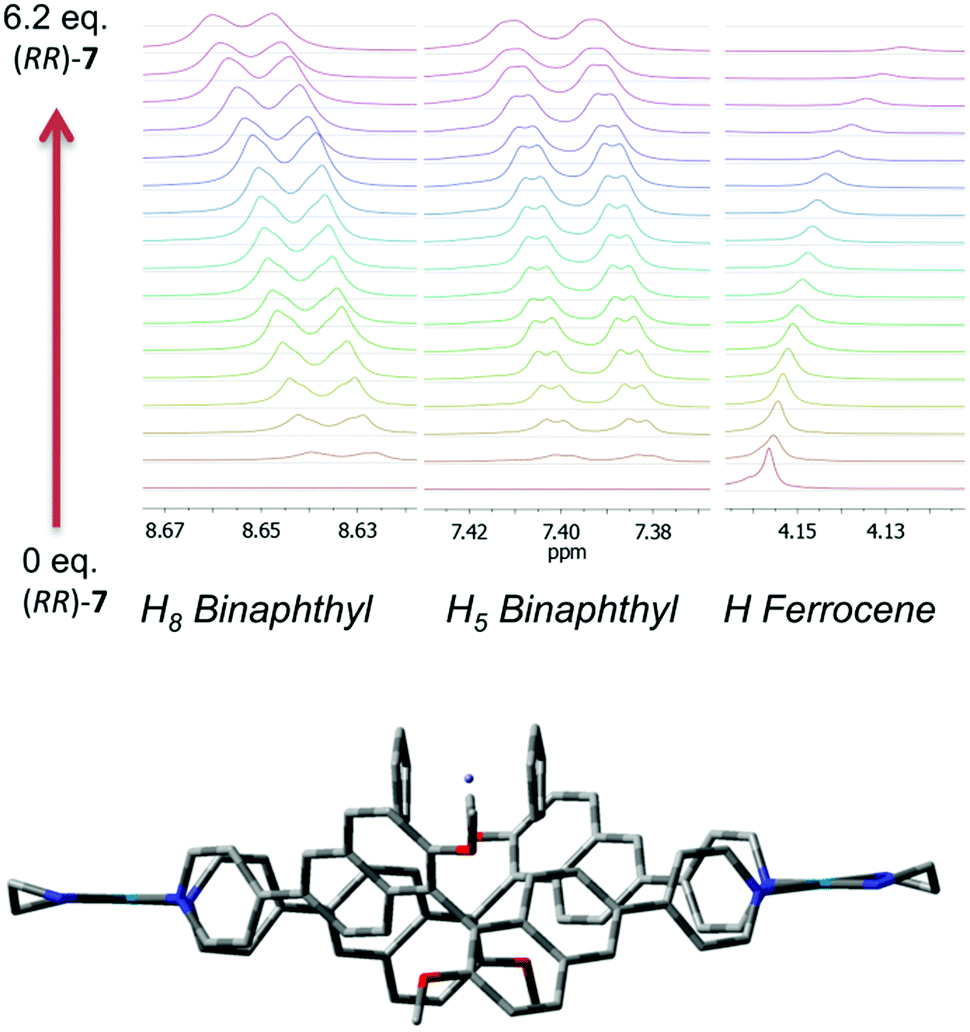 | ||
| Fig. 3 Top: Different regions of the 1H NMR spectra (500 MHz, CD3CN) showing the titration of ferrocene (2.4 mM) with increasing amounts of compound (RR)-7. Bottom: Molecular modelling of the 1:1 complex. | ||
Further UV/Vis and CD titrations on compound (RR)-7 were carried out in different solvent mixtures (MeCN:H2O in different ratios such as 100:0, 90:10, 70:30, Fig. S5, ESI†) to gain further insights into the binding mechanisms, especially in terms of solvophobic interactions. In order to adequately delineate the thermodynamic association constants and the stoichiometry of the supramolecular complexes, the entire set of wavelengths was modelled simultaneously using a non-linear least-squares regression program for performing equilibrium-restricted factor analysis.8 The model for the best fit of all UV titration data needed to include both 1:1 and 1:2 macrocycle:ferrocene complex stoichiometries (Tables S1–S4, ESI†). Calculated values for the binding in pure MeCN were logK11 = 4.4 kcal mol−1 and log K12 = 2.7 kcal mol−1. Although some changes could be observed in the distribution between the 1:1 and 1:2 complexes upon changing the solvent mixture, the changes in the total binding energy are rather limited, suggesting that contrasting noncovalent interactions (including dipole–dipole interactions) play a role in the binding, and that net hydrophobic and/or solvent polarity effects cannot be considered exclusively. Modelling the data obtained from the CD titration in MeCN confirmed the presence of both 1:1 and 1:2 host:guest complexes as well as the values for the binding constants calculated using the UV/Vis data (Table S5, ESI†).
Further confirmations about the nature and the stoichiometry of the species in solution came from 1H NMR titrations and cyclic voltammetry experiments. Best results by 1H NMR were obtained by titrating a solution of ferrocene in d3-MeCN at constant concentration with increasing amounts of macrocycle (Fig. 3, top, Fig. S6–S11 in the ESI†). In this case, small but measurable shifts were observed not only in the ferrocene proton resonances, but also for selected proton resonances of the macrocycle which are the closest contacts in the minimized model of the complex.
NMR shifts for the ferrocene protons could be fitted (Table S5, ESI†) with a high confidence using a 1:2 host:guest model, giving a negligible contribution for logK11 and a value for logK12 = 4.8 kcal mol−1.||
The optimized molecular modelling of the 1:1 complex (Fig. 3, bottom) showed the guest sitting on the top of the host rather than buried inside its molecular cavity. The distance of the closest ferrocene hydrogens from binaphthyl units is in the range of 3–4 Å. The dihedral angle between the naphthyl planes of the binaphthyl units is 83°, virtually the same as that measured for the molecular modelling of (RR)-7 alone. For the 1:2 complex (Fig. S12, ESI†) two isomers were found that differ by less than 3 kcal mol−1. The alignment of the main axis of both ferrocene molecules is parallel to that formed by palladium atoms. The affinity of ferrocene with (RR)-7 could also be monitored using cyclic voltammetry in solution. Again, a shift in the oxidation and reduction potential of the Fc/Fc+ couple could be observed, reaching a maximum of 40 mV in the reduction mode when a macrocycle:ferrocene stoichiometry of 1:2 is reached (Fig. S13, ESI†).
Encouraged by the much larger response using CD with respect to the UV/Vis techniques in binding ferrocene, we performed experiments in the presence of a potentially interfering analyte, 2-hydroxy-4-methoxy-benzophenone, with a distinct absorbance band (λmax = 320 nm, ε = 105 M−1 cm−1), right in the UV/Vis detection region of our complex. The titrations reported in Fig. 2 were thus repeated by adding to the host a concentrated solution of the benzophenone as a potentially interfering analyte. In the UV/Vis experiment, the benzophenone absorbance adds to the decreasing band of the complex in a disruptive manner, so that the differences between the detection carried out with and without the interferent are not marginal (Fig. 4, left). In the case of the CD titration, instead, the interferent is CD silent, and the results with or without the interferent are essentially superimposable (Fig. 4, right).
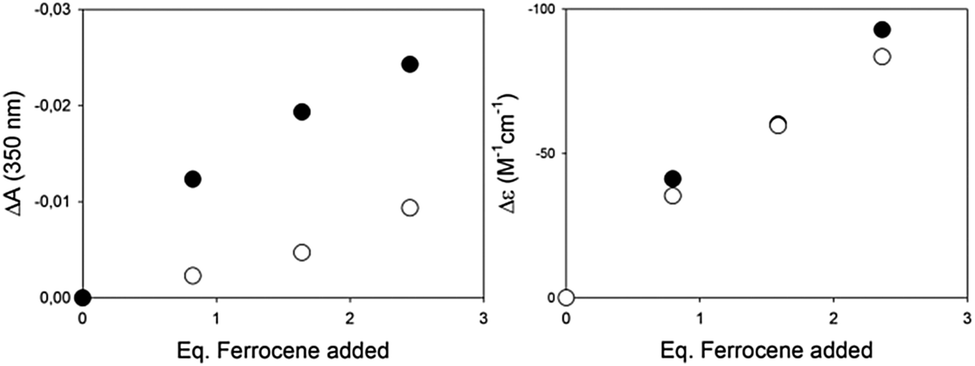 | ||
| Fig. 4 Left: Differences in the UV/Vis absorbance at 350 nm upon titration of (RR)-7 (1 × 10−5 M in MeCN) with ferrocene, in the absence (filled dots) or in the presence (empty dots) of 2-hydroxy-4-methoxy-benzophenone (7 eq. vs. host, 7 × 10−5 M). Right: Differences in the CD output (272 nm) upon titration of (RR)-7 (6 × 10−6 M in MeCN) with ferrocene in the absence (filled dots) or in the presence (empty dots) of 2-hydroxy-4-methoxy-benzophenone (14 eq. vs. host, 8.5 × 10−5 M). | ||
Rather than the host binding selectively one of multiple guests, the competitive experiment highlights the potential of the chiroptical readout when other readout techniques (UV/Vis absorbance in our case) are hampered by the presence of an interfering analyte.
In conclusion, we have presented the first example of host–guest chemistry related to ferrocene11 in which detection is chiroptical. The metal-linked macrocycle is formed in high yields and with a geometry capable of size selective, strong binding of ferrocene. Given the complementarity of the CD and UV/Vis response, our molecular sensor has potential in the detection of ferrocene in complex analytical matrices. In light of the continuing importance and use of ferrocene and its derivatives in materials science,12 our discovery may open new avenues in the design of functional organic materials and sensors through supramolecular chemistry.
DP acknowledges support from MIUR (Programs of National Relevant Interest PRIN grant 2009-A5Y3N9) and, in part, from INSTM-Regione Lombardia (2010–2012 and 2013–2015). DVG acknowledges support from the National Science Foundation (grant number CHE-1310402). Additionally, availability and use of the 500 MHz NMR instrument of the NMR Lab, NCSR “Demokritos”, Greece, a member of INSTRUCT-EL, is gratefully acknowledged.
Notes and references
- (a) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef PubMed; (b) J. W. Canary, S. Mortezaei and J. Liang, Coord. Chem. Rev., 2010, 254, 2249–2266 CrossRef; (c) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef PubMed.
- (a) G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2011, 40, 4603–4625 RSC; (b) D. Pasini and A. Nitti, Chirality, 2016, 28, 116–123 CrossRef PubMed; (c) M. Anyika, H. Gholami, K. D. Ashtekar, R. Acho and B. Borhan, J. Am. Chem. Soc., 2014, 136, 550–553 CrossRef PubMed; (d) K. W. Bentley and C. Wolf, J. Am. Chem. Soc., 2013, 135, 12200–12203 CrossRef PubMed; (e) S. Superchi, D. Casarini, A. Laurita, A. Bavoso and C. Rosini, Angew. Chem., Int. Ed., 2001, 40, 451–454 CrossRef; (f) M. Caricato, C. Coluccini, D. Dondi, D. A. Vander Griend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 RSC; (g) M. Caricato, N. J. Leza, K. Roy, D. Dondi, G. Gattuso, L. S. Shimizu, D. A. Vander Griend and D. Pasini, Eur. J. Org. Chem., 2013, 6078–6083 CrossRef; (h) M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef; (i) C. Rosini, S. Superchi, H. W. I. Peerlings and E. W. Meijer, Eur. J. Org. Chem., 2000, 61–71 CrossRef.
- (a) L. Xu, M. Sun, W. Ma, H. Kuang and C. Xu, Mater. Today, 2016 DOI:10.1016/j.mattod.2016.05.015; (b) W. Ma, H. Kuang, L. Xu, L. Ding, C. Xu, L. Wang and N. A. Kotov, Nat. Commun., 2013, 4, 2689 Search PubMed.
- M. Caricato, A. K. Sharma, C. Coluccini and D. Pasini, Nanoscale, 2014, 6, 7165 RSC.
- (a) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef PubMed; (b) J. K. Klosterman, Y. Yamauchi and M. Fujita, Chem. Soc. Rev., 2009, 38, 1714 RSC.
- (a) M. Fujita, S. Nagao, M. Iida, K. Ogata and K. Ogura, J. Am. Chem. Soc., 1993, 115, 1574 CrossRef CAS; (b) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- (a) J. Bunzen, T. Bruhn, G. Bringmann and A. Lützen, J. Am. Chem. Soc., 2009, 131, 3621–3630 CrossRef CAS PubMed; (b) S. J. Lee and W. Lin, Acc. Chem. Res., 2008, 41, 521 CrossRef CAS PubMed.
- http://www.calvin.edu/~dav4/Sivvu.htm. See also: D. A. Vander Griend, D. K. Bediako, M. J. DeVries, N. A. DeJong and L. P. Heeringa, Inorg. Chem., 2008, 47, 656–662 CrossRef CAS PubMed.
- S. J. Lee, J. S. Kim and W. Lin, Inorg. Chem., 2004, 43, 6579 CrossRef CAS PubMed.
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- For recent examples of macrocyclic receptors for ferrocene, see: J. J. Henkelis, A. K. Blackburn, E. J. Dale, N. A. Vermeulen, M. S. Nassar and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 13252–13255 CrossRef CAS PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Overall synthesis, methods and experimental procedures, and additional titration studies. See DOI: 10.1039/c6cc05937f |
| ‡ Current address: Institute of Nanoscience & Nanotechnology, NCSR “Demokritos”, Terma Patr. Gregoriou & Neapoleos, Aghia Paraskevi, 15310, Attikis, Greece. |
| § By using an analogue to compound (R)-5 bearing ethoxy instead of methoxy substituents in the 2,2′ positions of the BINOL skeleton, D4 symmetrical cyclic tetramers, as the only products, using Pd2+-based corners, were obtained. See ref. 9. |
| ¶ Structure optimization was performed using the semi-empirical PM3 method with force fields present in the Hyperchem package. |
| || The discrepancy between thermodynamic values obtained by UV/Vis and NMR titrations in MeCN, obtained at different concentrations, may be rationalized by the presence of the soft counteranions, which can play a role with their ion pairing equilibria, and bring additional variables in such a dynamic situation. See ref. 10 for related examples. |
| This journal is © The Royal Society of Chemistry 2016 |