
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of steric hindrance in the intramolecular oxidative aromatic coupling of pyrrolo[3,2-b]pyrroles†
Maciej
Krzeszewski
a,
Paweł
Świder
a,
Łukasz
Dobrzycki
b,
Michał K.
Cyrański
*b,
Witold
Danikiewicz
*a and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl; witold.danikiewicz@icho.edu.pl
bWarsaw University, Chemistry Department, Pasteura 1, 01-224 Warsaw, Poland. E-mail: chamis@chem.uw.edu.pl
First published on 25th August 2016
Abstract
The presence of steric hindrance triggers different reaction pathways in the intramolecular oxidative aromatic coupling of tetraaryl-pyrrolo[3,2-b]pyrroles and leads to the formation of a fluorene moiety and a new cationic π-system linked together by a spiro carbon atom. Computational studies elegantly rationalized these results. These previously unknown functional dyes emit red light with reasonable efficiency.
Oxidative aromatic coupling and the Scholl reaction are often the methods of choice in the synthesis of large π-extended scaffolds.1 Indeed, in some cases over 100 C–C bonds are formed from suitable precursors furnishing truly amazing polycyclic aromatic hydrocarbons in one synthetic operation.2 Yet despite being known for almost 150 years, the oxidative aromatic coupling reaction continues to surprise chemists.3 The breakthrough works of various groups,4 most prominently Müllen5 and Durola,6 have proven that there is often no obvious relationship between steric hindrance and the output of this reaction. A general set of rules which would predict when and how this reaction occurs is still beyond our current reach. Additionally, the mechanistic aspects are still under discussion,1a,7 although Waldvogel and co-workers have recently made significant progress.8
The extraordinarily easy access to 1,2,4,5-tetraaryl-pyrrolo[3,2-b]pyrroles9 and their intrinsic electron-rich core make them ideal building blocks for the study on oxidative aromatic coupling.10 Our examination of the relationship between the structure of aromatic compounds and the results of oxidative aromatic coupling has provided key information for truly understanding this system as well as the discovery of a new fluorophore. Herein we present the results of this study.
We began our investigation with the preparation of the corresponding 2-arylbenzaldehydes 1b–f using Suzuki–Miyaura coupling. In order to obtain aldehydes 1b–c and 1e–f, respective bromides (iodides) were reacted with 2-formylphenylboronic acid. To synthesize derivative 1d, 2-bromobenzaldehyde was reacted with 3,5-dimethylphenylboronic acid. Aldehydes 1b–f were synthesized in 60–98% yields (see the ESI†), and they were subsequently used in the synthesis of tetraaryl-pyrrolo[3,2-b]pyrroles, a procedure developed and optimized in our laboratory (Scheme 1).9b Desired products 4a–e were smoothly obtained in yields ranging from 34% to 48% (Scheme 1 and Table 1). The only exception was the reaction of electron-rich aldehyde 3f. The expected compound 4f (based on ESI-MS of the crude reaction mixture) did not form and only tarry products were observed. The most plausible reason behind this result is the low intrinsic stability of hypothetical compound 4f possessing an electron-rich pyrrolo[3,2-b]pyrrole core decorated with electron-donating substituents. Most likely, the oxidation potential of 4f is low enough to facilitate its immediate transformation into polymeric materials in the presence of air.
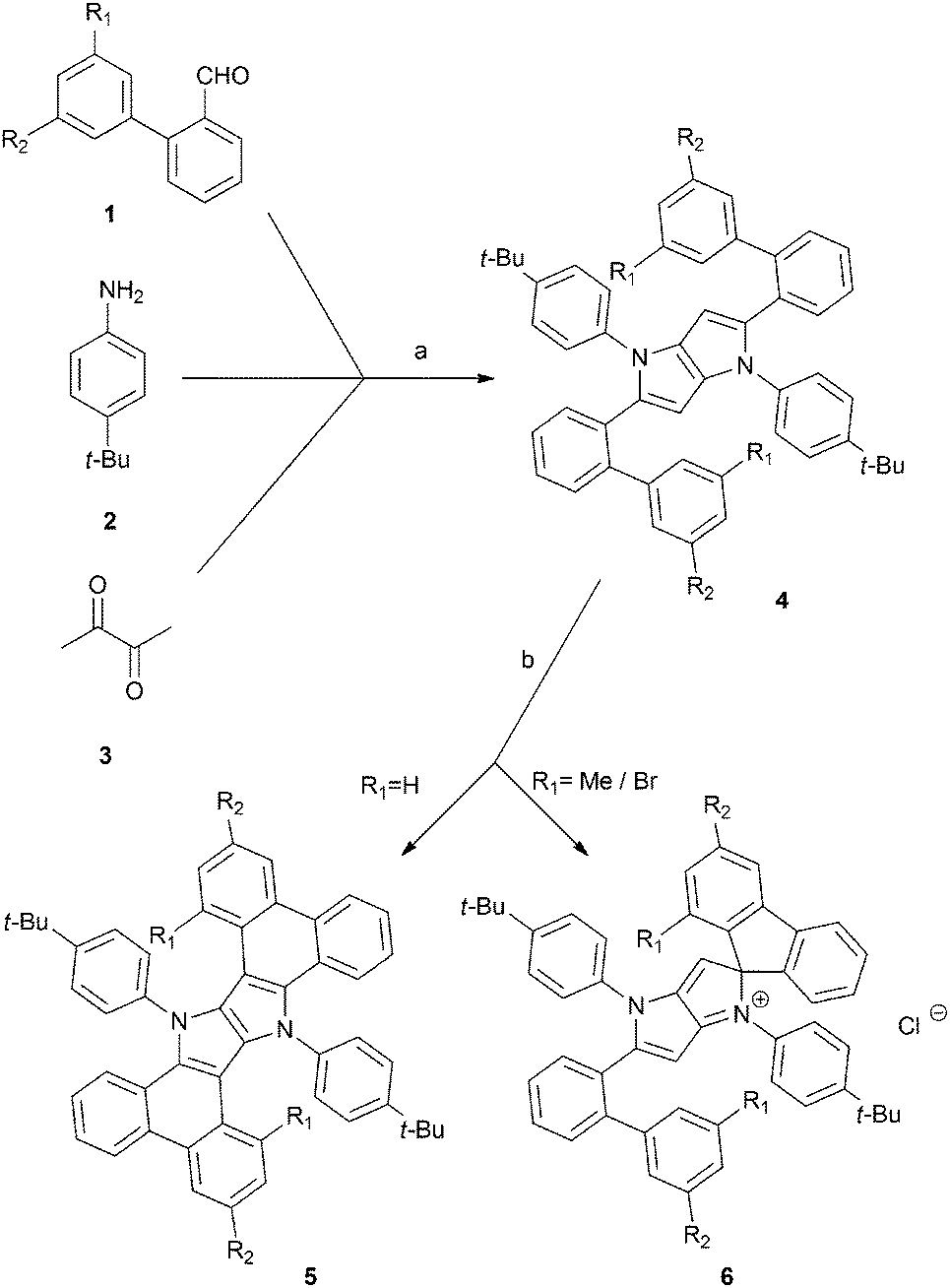 | ||
| Scheme 1 General method for the synthesis of π-expanded compounds 5 and spiro-derivatives 6. Conditions: (a) TsOH, AcOH, 90 °C, 3 h, (b) FeCl3, MeNO2, DCM, r.t., 30 min. | ||
In accordance with our previous results,10 we subjected 4b–4e to FeCl3 in MeNO2/CH2Cl2. In the case of substrates 4b and 4c, ladder-type π-expanded pyrrolo[3,2-b]pyrroles 5b and 5c were formed according to our expectations via double intramolecular oxidative aromatic coupling (Scheme 1 and Table 1). On the other hand, unexpected results were observed for more congested compounds. Derivatives 4d (R1 = R2 = Me) and 4e (R1 = R2 = Br) were reacted with iron(III) chloride and produced very polar red products. Analysis of their NMR spectra led to inconclusive results and eventually the structure of compound 6e was verified via X-ray crystallography. The single crystal structure of compound 6e was established based on the procedures described in the references given in the ESI.† To our surprise the structure of compound 6e contained a spiro carbon atom (Fig. 1).
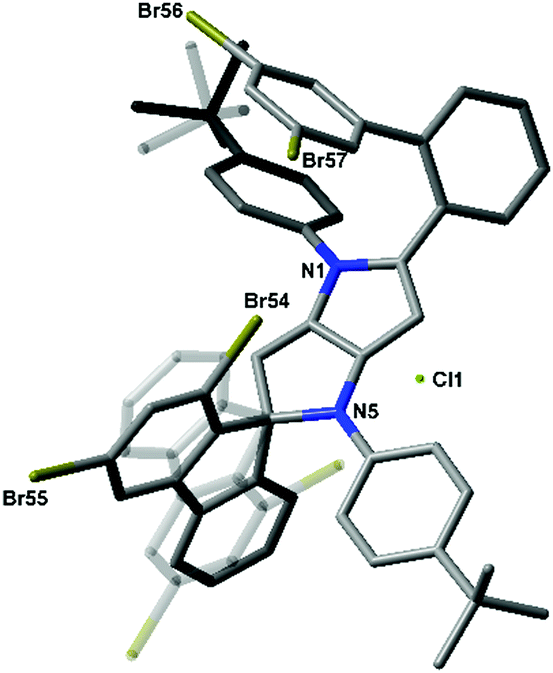 | ||
| Fig. 1 Molecular structure of 6e. Disordered fluorene and t-butyl fragments with lower occupancies are made transparent. H atoms and the disordered Et2O molecule are omitted for clarity. CCDC 1487685. | ||
The structures of 4d and 4e differ from their analogues 4b and 4c in that they possess two substituents (Me or Br) adjacent to the position of the expected C–C bond forming process (Scheme 1). The structures of salts 6d and 6e made it clear that the radical cation formed on the electron-rich pyrrolopyrrole moiety attacked the aryl substituent in a different way, forming a C–C bond on an already occupied position (rather than unoccupied position 3). Consequently, this led to the formation of a spiro system and the new chromophore (Scheme 1 and Fig. 1). The new core being effectively cation, in contrast to compounds 4, is electron-deficient. Hence the reaction did not occur any further from the second site of the molecule. Interestingly, these unforeseen spiro derivatives 6d and 6e formed in almost quantitative yields (Table 1). It is noteworthy to add that recently Waldvogel and co-workers revealed the formation of spirocyclic compounds via intramolecular oxidative coupling of cinnamates.11
To rationalize why the oxidation of compounds 4a–4c results in the formation of six-membered rings while in the case of compounds 4d and 4e spiro five-membered rings are formed, we decided to model both reactions using quantum chemical calculations. Due to the relatively large size of the studied molecules Truhlar's unrestricted M05-2X DFT functional with the 6-31G(d) basis set has been selected as the method of choice.12 Compounds 4a and 4e with removed t-butyl groups have been selected as the representative models. Calculations were performed using the Gaussian 09 rev. A.02 software package.13 Full computational details, including Cartesian coordinates of all calculated structures, are available in the ESI.†
As the starting point we assumed that the reaction starts with the formation of a radical cation of the substrate, which subsequently can undergo cyclization through different transition states (TS), resulting in the formation of either a 6-membered ring or a spiro-5-membered ring (Scheme 2). In the next step these intermediates are oxidized, yielding final products: a neutral molecule in the first case and a cation in the second one. The results of the calculations are summarized in Fig. 2. In the case of compound 7a (an analogue of 4a without t-butyl groups) both the activation energy barriers and the stability of the products favor the formation of a 6-membered ring. A different situation has been observed for tetrabromosubstituted compound 7e. In this case the formation of a spiro-5-membered ring is preferred by both TS energy and the relative energy of the product. Both results are in full agreement with the experiments. These results also revealed that it is not possible to tell if the reaction is controlled kinetically or thermodynamically because in both cases the kinetic and thermodynamic products are the same. The main reason for the different reactivities of compounds 4a–c and 4d–e is the steric hindrance. Examination of the calculated structures of compounds 7a and 7e shows that in the case of the latter the formation of the transition state leading to the spiro 5-membered ring is less sterically demanding. A rather surprising observation is that the activation energy of the formation of both transition states for compound 7e is lower than in the case of compound 7a, which is obviously less sterically crowded. A plausible rationalization for this effect is that compound 7e is already sterically crowded so the difference in steric hindrance between the starting molecule and TS is lower than for compound 7a.
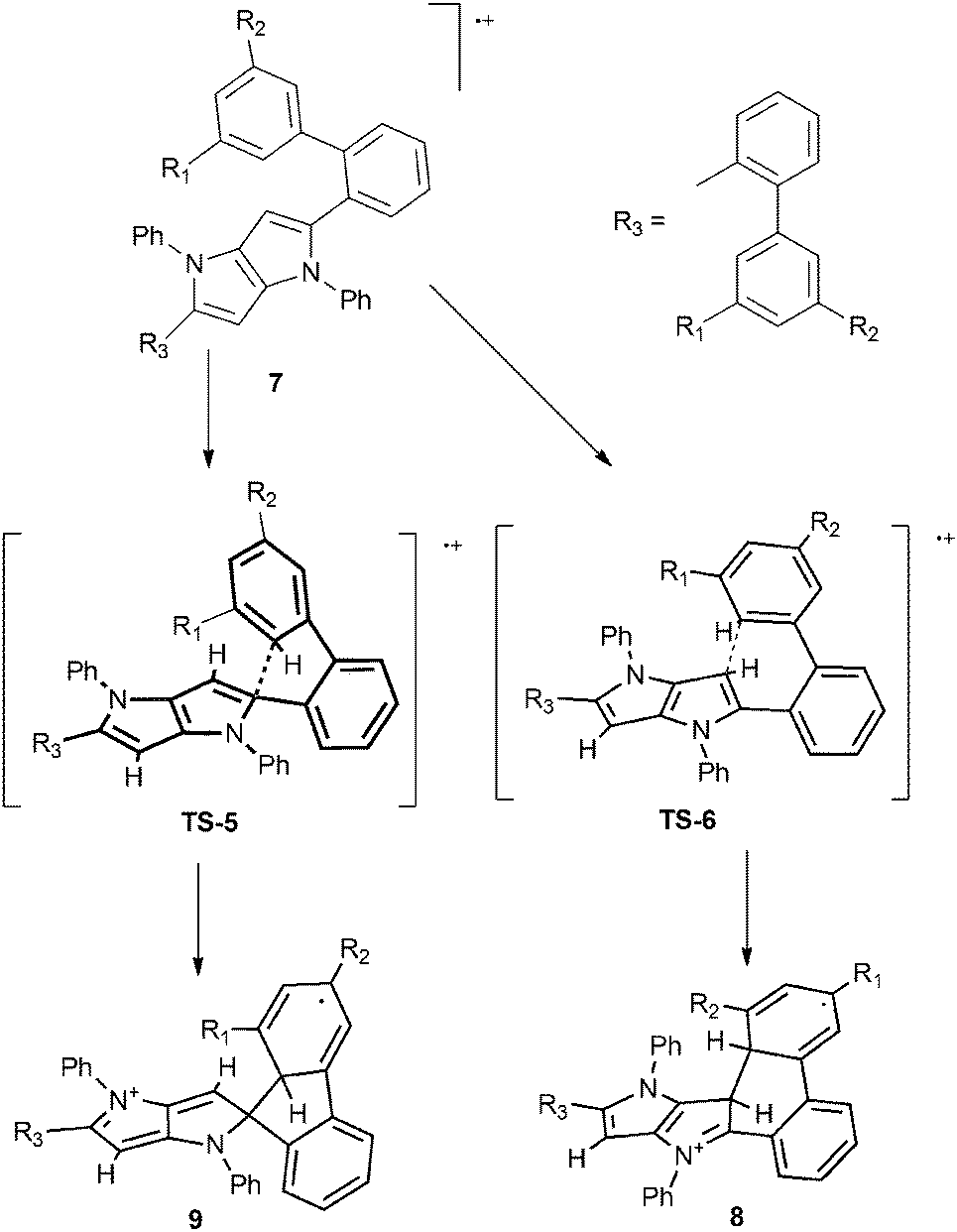 | ||
| Scheme 2 Proposed reaction mechanism leading to the formation of the 6-membered ring (8) and the spiro-5-membered ring (9). | ||
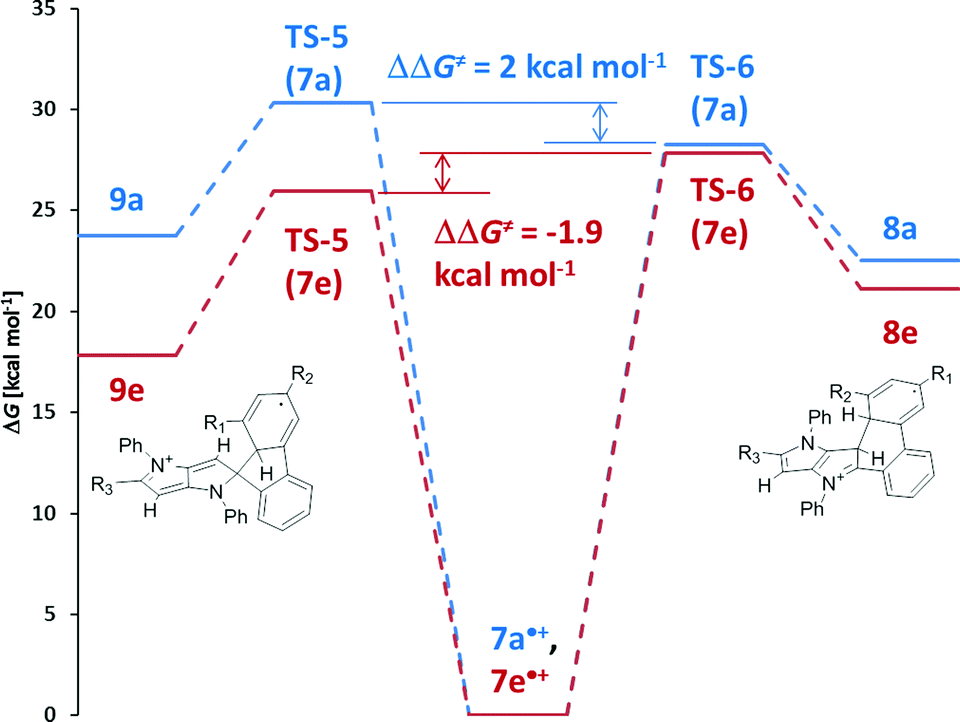 | ||
| Fig. 2 Gibbs free energy diagrams of the reactions of radical cations of compounds 7a (blue) and 7e (red) in the gas phase leading to the formation of the 6-membered ring (right side) and the spiro-5-membered ring (left side). | ||
The optical properties of compounds 4b–e and 5b–c were analogous to previously described 4a and 5a (Fig. 3–5 and Table 2).10 The untypical direction of intramolecular oxidative coupling in the case of 4d and 4e led to the formation of not only a new heterocyclic scaffold but also a new dye. Consequently, both absorption and emission of compounds 6d and 6e were entirely different from those of 5b–c (Fig. 4 and 5 and Table 2). The absorption maxima were in the green region of the spectra. Both compounds emitted red light (λem 595–606 nm) with moderate intensity.
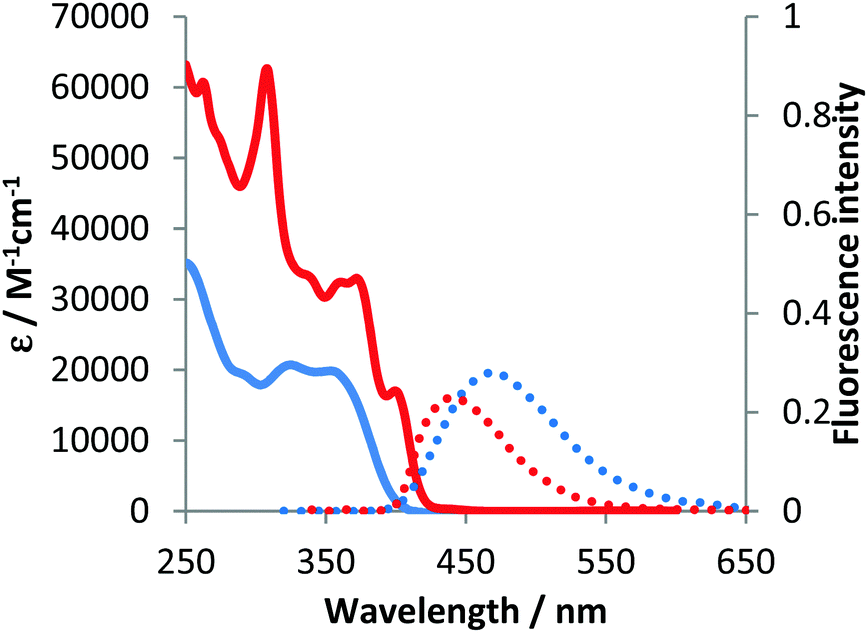 | ||
| Fig. 3 Absorption (solid) and emission (dotted) spectra of 4b (blue line) and 5b (red line) measured in dichloromethane. | ||
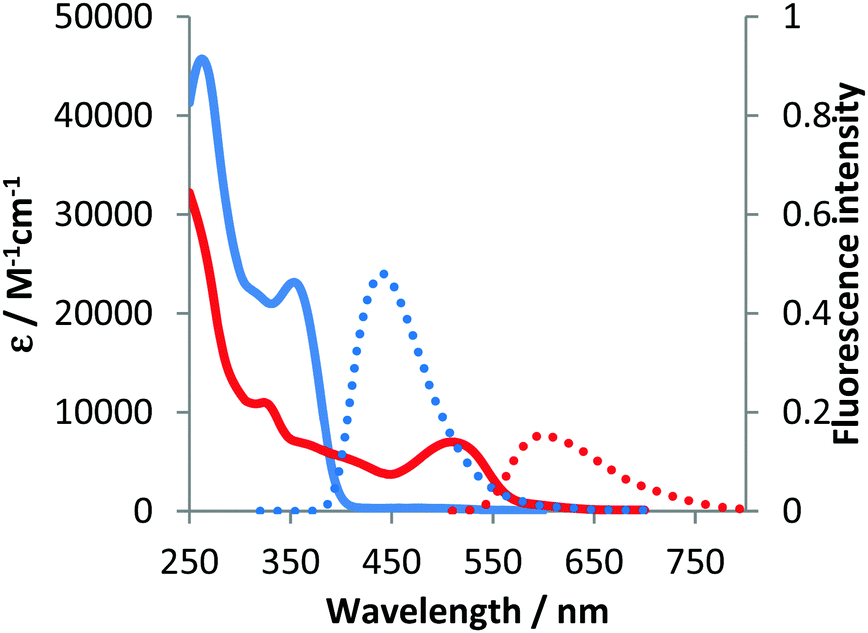 | ||
| Fig. 4 Absorption (solid) and emission (dotted) spectra of 4d (blue line) and 6d (red line) measured in dichloromethane. | ||
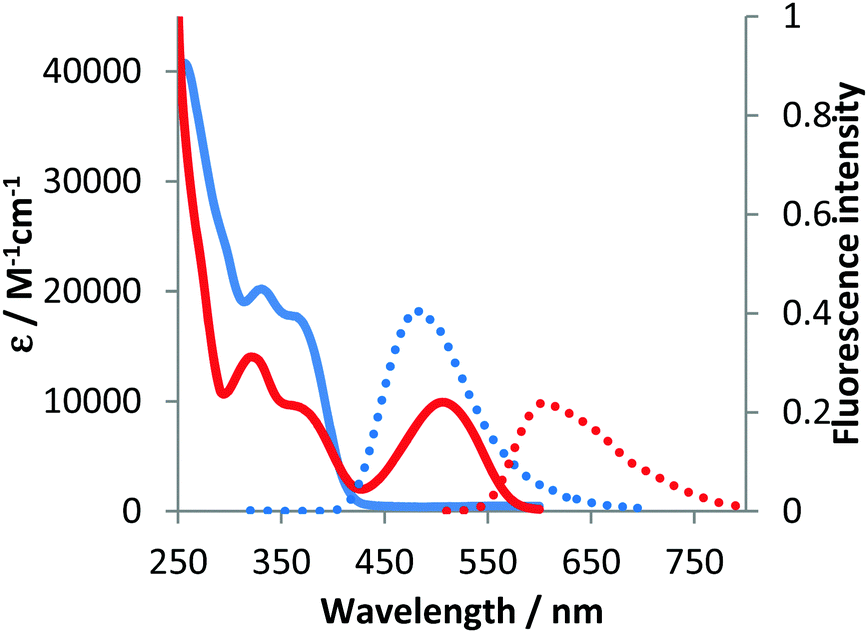 | ||
| Fig. 5 Absorption (solid) and emission (dotted) spectra of 4e (blue line) and 6e (red line) measured in dichloromethane. | ||
| Cmpd. | λ abs [nm] | λ em [nm] | Stokes shift [cm−1] | ε max [M−1 cm−1] | Φ fl |
|---|---|---|---|---|---|
| a Determined with quinine sulfate in H2SO4 (0.5 M) as a standard. b Ref. 10. c Spectra measured in toluene. d Determined with Rhodamine 6G in ethanol as a standard. | |||||
| 4a | 350 | 452 | 6400 | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.84 |
| 4b | 353 | 461 | 6600 | 20000 |
0.38 |
| 4c | 353 | 428 | 5000 | 22000 |
0.37 |
| 4d | 353 | 441 | 5600 | 23000 |
0.62 |
| 4e | 364 | 481 | 6700 | 18000 |
0.13 |
| 5a | 372 | 428 | 3500 | 39000 |
0.45 |
| 5b | 399 | 444 | 2500 | 17000 |
0.02 |
| 5c | 405 | 415 | 600 | 21500 |
0.35 |
| 6d | 508 | 595 | 2900 | 7000 | 0.34d |
| 6e | 506 | 606 | 3300 | 10000 |
0.22d |
The Stokes shift for these new dyes was moderate, which probably stems from the difference in the geometry of these molecules between the ground and the excited state. The bathochromically shifted absorption and emission are not apparent in these cases since the novel chromophore is rather small. Due to the steric hindrance the conjugation of the phenyl substituent located at position 5 is most probably minimal (in the ground state).
In conclusion, we have proven that large substituents present at positions adjacent to the site of oxidative aromatic coupling can completely alter the expected reaction pathway. In particular, C–C bonds can form on the already occupied quaternary carbon atom leading to the formation of a five-membered ring and consequently the spiro-product possessing a novel π-conjugated core, the existence of which was unambiguously confirmed by X-ray crystallography. The difference in the course of these two reactions has been post factum rationalized by calculating the thermodynamic stabilization energies of the intermediate structures. As a result of a change of the π-electron system, the absorption and emission of these small dyes are significantly bathochromically shifted versus tetraaryl-pyrrolo[3,2-b]pyrroles or their π-expanded analogues. This unexpected result adds an important piece of information towards understanding the relationship between the structure of the polycyclic aromatic compound and the results of its interaction with oxidants.
We thank the Foundation for Polish Science (MISTRZ programme), the Polish National Research Center (PRELUDIUM 2015/19/N/ST5/00826) and the Global Research Laboratory Program (2014K1A1A2064569) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning, Korea. M. K. thanks Yevgen Poronik for insightful consultations and Jillian Larsen (UC Riverside) for amending the manuscript.
Notes and references
- (a) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed; (b) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed; (c) J. He, S. Mathew, Z. J. Kinney, R. M. Warrell, J. S. Molina and C. S. Hartley, Chem. Commun., 2015, 51, 7245 RSC; (d) N. L. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30 CrossRef CAS PubMed; (e) N. Ono, H. Yamada and T. Okujima, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2009, vol. 2, pp. 1–102 Search PubMed; (f) A. N. Lakshminarayana, J. Chang, J. Luo, B. Zheng, K.-W. Huang and C. Chi, Chem. Commun., 2015, 51, 3604 RSC; (g) H. Zhong, C.-H. Wu, C.-Z. Li, J. Carpenter, C.-C. Chueh, J.-Y. Chen, H. Ade and A. K.-Y. Jen, Adv. Mater., 2016, 28, 951 CrossRef CAS PubMed; (h) J. Luo, X. Xu, R. Mao and Q. Miao, J. Am. Chem. Soc., 2012, 134, 13796 CrossRef CAS PubMed; (i) C.-M. Chou, S. Saito and S. Yamaguchi, Org. Lett., 2014, 16, 2868 CrossRef CAS PubMed; (j) J. He, D. M. Agra-Kooijman, G. Singh, C. Wang, C. Dugger, J. Zeng, L. Zang, S. Kumar and C. S. Hartley, J. Mater. Chem. C, 2013, 1, 5833 RSC; (k) B. Kramer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2004, 43, 2446 CrossRef CAS PubMed; (l) B. Koszarna and D. T. Gryko, Chem. Commun., 2007, 2994 RSC; (m) S. Ooi, T. Tanaka, K. H. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 6535 CrossRef CAS PubMed.
- (a) A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126 CrossRef CAS PubMed; (b) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS PubMed.
- (a) N. Fukui, S.-K. Lee, K. Kato, D. Shimizu, T. Tanaka, S. Lee, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2016, 7, 4059 RSC; (b) J. He, J. L. Crase, S. H. Wadumethrige, K. Thakur, L. Dai, S. Zou, R. Rathore and C. S. Hartley, J. Am. Chem. Soc., 2010, 132, 13848 CrossRef CAS PubMed; (c) S. L. Skraba-Joiner, E. C. McLaughlin, A. Ajaz, R. Thamatam and R. P. Johnson, J. Org. Chem., 2015, 80, 9578 CrossRef CAS PubMed.
- (a) M. Danz and G. Hilt, Adv. Synth. Catal., 2011, 353, 303 CrossRef CAS; (b) B. T. King, J. Kroulik, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, J. Org. Chem., 2007, 72, 2279 CrossRef CAS PubMed.
- (a) J. Liu, A. Narita, S. Osella, W. Zhang, D. Schollmeyer, D. Beljonne, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 2602 CrossRef CAS PubMed; (b) L. Dössel, L. Gherghel, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2540 CrossRef PubMed.
- A. Pradhan, P. Dechambenoit, H. Bock and F. Durola, Angew. Chem., Int. Ed., 2011, 50, 12582 CrossRef CAS PubMed.
- (a) P. Rempala, J. Kroulik and B. T. King, J. Am. Chem. Soc., 2004, 126, 15002 CrossRef CAS PubMed; (b) N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848 CrossRef CAS PubMed; (c) P. Rempala, J. Kroulik and B. T. King, J. Org. Chem., 2006, 71, 5067 CrossRef CAS PubMed.
- (a) K. Wehming, M. Schubert, G. Schnakenburg and S. R. Waldvogel, Chem. – Eur. J., 2014, 20, 12463 CrossRef CAS PubMed; (b) M. Schubert, S. Trosien, L. Schulz, C. Brandscheid, D. Schollmeyer and S. R. Waldvogel, Eur. J. Org. Chem., 2014, 7091 CrossRef CAS; (c) J. Leppin, M. Schubert, S. R. Waldvogel and K. Heinze, Chem. – Eur. J., 2015, 21, 4229 CrossRef CAS PubMed; (d) M. Schubert, P. Franzmann, A. Wünsche von Leupoldt, K. Koszinowski, K. Heinze and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 1156 CrossRef CAS PubMed.
- (a) A. Janiga, E. Głodkowska-Mrówka, T. Stokłosa and D. T. Gryko, Asian J. Org. Chem., 2013, 2, 411 CrossRef CAS; (b) M. Krzeszewski, B. Thorsted, J. Brewer and D. T. Gryko, J. Org. Chem., 2014, 79, 3119 CrossRef CAS PubMed; (c) A. Janiga, M. Krzeszewski and D. T. Gryko, Chem. – Asian J., 2015, 10, 212 CrossRef CAS PubMed.
- M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2015, 80, 2893 CrossRef CAS PubMed.
- M. Schubert, K. Wehming, A. Kehl, M. Nieger, G. Schnakenburg, R. Frohlich, D. Schollmeyer and S. R. Waldvogel, Eur. J. Org. Chem., 2016, 60 CrossRef CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed, for full citation see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR and 13C NMR spectra, UV-Vis absorption and emission spectra, details of DFT calculations and data on the crystal structure of compound (6f). CCDC 1487685. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc05904j |
| This journal is © The Royal Society of Chemistry 2016 |