
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelix–helix inversion of an optically-inactive π-conjugated foldamer triggered by concentration changes of a single enantiomeric guest leading to a change in the helical stability†
Lijia
Liu
ab,
Naoki
Ousaka
*a,
Miki
Horie
a,
Fumihiko
Mamiya
a and
Eiji
Yashima
*a
aDepartment of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: ousaka@apchem.nagoya-u.ac.jp; yashima@apchem.nagoya-u.ac.jp; Fax: +81 52-789-3185
bPolymer Materials Research Center, Key Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin, 150001, China
First published on 18th August 2016
Abstract
A preferred-handed helicity induced in an optically-inactive poly(phenyleneethynylene)-based foldamer bearing carboxylic acid pendants upon complexation with a single enantiomeric diamine was subsequently inverted into the opposite helix upon further addition of the diamine, accompanied by a remarkable change in the stability of the helices.
Stimuli-triggered switching or inversion of macromolecular and supramolecular helicity including a foldamer in a controllable manner has become a fascinating research area of polymer and supramolecular chemistry in the last two decades, because of potential application to chiral materials for asymmetric catalysis, chiral sensing and chiral separation.1 Among them, the inversion of the helicity in optically active helical systems has been successfully achieved via various external stimuli, such as temperature,2 solvent,1g,2g,3 photo-irradiation4 and chiral2g,5 or achiral additives.6
Previously, we reported a thermo- or solvent-driven helicity inversion of a preferred-handed helical polymer induced by only a single enantiomer through noncovalent interactions in a dynamically racemic helical poly(phenylacetylene) bearing phosphonate pendants.7 However, there are only a few examples of such a noncovalent approach to produce diastereomeric right- and left-handed helices from such racemic helices upon complexation with a non-racemic guest.7,8 We now report a unique helicity induction and subsequent inversion of the helicity in an optically inactive poly(phenyleneethynylene) (PPE)-based foldamer1c,9 composed of alternating achiral o-diethynylphthalic acid and p-phenyleneethynylene (PE) units (poly-1) upon complexation with the increasing amount of a single enantiomeric diamine (A1), thus producing pseudo-diastereomeric right- and left-handed helices (Fig. 1). Interestingly, the stability of the diastereomeric helices was significantly dependent on the helical senses induced by different amounts of the chiral diamine. An analogous optically-active PPE-based foldamer bearing chiral substituents (poly-2) was also prepared for comparison.
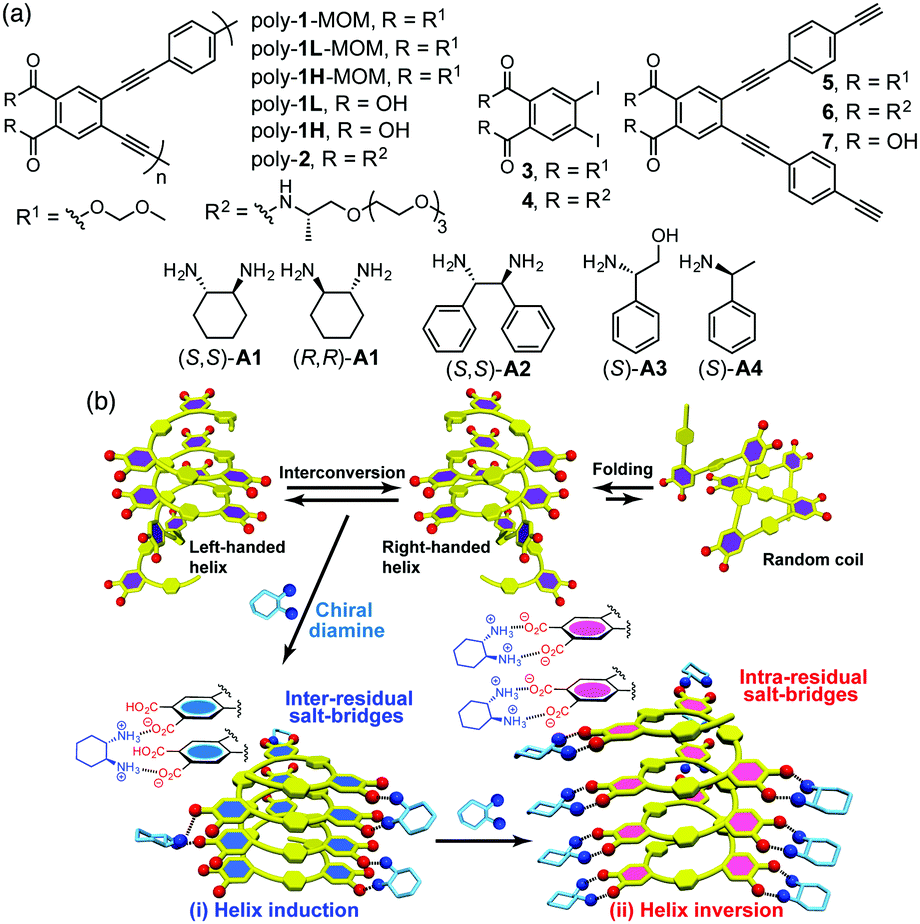 | ||
| Fig. 1 (a) Chemical structures of poly(phenyleneethynylene) derivatives, monomers, and chiral amines. L, H, and MOM represent low and higher molecular weight parts of the fractionated poly-1-MOM or poly-1, and methoxymethyl, respectively. (b) Schematic representation of a preferred-handed helicity induction in an optically inactive poly-1 with a small amount of (S,S)-A1 (i) and subsequent inversion of the induced helicity upon further addition of (S,S)-A1 (ii). The plausible interaction modes are also depicted. | ||
The optically-inactive and active (3–7) monomers and polymers (poly-1 and poly-2) (Fig. 1a) were prepared according to Scheme S1 (ESI†). The achiral PPE-based foldamer with methoxymethyl (MOM) protecting groups (poly-1-MOM) was synthesized by the copolymerization of o-diiodo benzene 3 with the diethynyl monomer 5 using the palladium/copper-catalyzed Sonogashira cross-coupling reaction, producing a poly(o-PE-alt-p-PE)-based foldamer poly-1-MOM in a high yield (84%). In order to investigate the chain-length effect of the polymer on the chiroptical properties, the resulting poly-1-MOM was fractionated into a higher molecular weight fraction (poly-1H-MOM, Mn = 6.7 × 103) and a low molecular weight fraction (poly-1L-MOM, Mn = 4.4 × 103) using size-exclusion chromatography (Table S1, ESI†), followed by deprotection of the MOM groups, affording amphiphilic poly-1H and poly-1L consisting of the hydrophobic PE backbone and hydrophilic carboxyl side chains (Fig. 1a). In a similar way, an amphiphilic, chiral-amide-appended PPE-based foldamer (poly-2, Mn = 9.9 × 103) was also prepared from the corresponding monomers 4 and 6 (Fig. 1a and Table S1, ESI†).
Because of the amphiphilic nature of poly-1, it is important to choose a suitable solvent system to induce the solvophobically-driven helical folding in the PE backbone.1c,9 Therefore, an H2O/THF mixture (6.5/3.5, v/v) was chosen as the solvent, because poly-1H and poly-1L are hardly soluble in pure H2O, which is a poor solvent for the PE backbone. The folding properties of poly-1L in the absence and presence of a series of enantiopure mono- (A3, A4) and diamines (A1, A2) were first investigated via a combination of circular dichroism (CD), UV-vis and fluorescence measurements, which is known to be a powerful method to obtain valuable information on the helical folding for analogous foldamers composed of o-PE,9c,do-PE-alt-p-PE,9f,g and m-PE1c,9a,b,e backbones.
In the presence of a small amount of (S,S)- and (R,R)-trans-1,2-cyclohexanediamine A1 (0.19 equiv. to the o-PE monomer unit), the optically inactive poly-1L exhibited perfect mirror-image induced CD (ICD) spectra in the H2O/THF mixture (ii and iv, Fig. 2a),10 whereas no ICD was observed for both poly-1L in DMSO and monomer 7 in H2O/THF (6.5/3.5, v/v) even with an excess of (S,S)-A1 (Fig. S1, ESI†). The CD spectral pattern induced in poly-1L with (R,R)-A1 is very similar to that of the optically-active poly-2 (Fig. S2, ESI†).11 In addition, we previously reported that an analogous oligomer composed of the same optically-active o-PE units folds into a right-handed helical conformation stabilized by intramolecular hydrogen bonds between the side-chain amide groups.12 These results suggest that poly-1L complexed with a small amount of (R,R)-A1 most likely takes the same right-handed helical conformation.
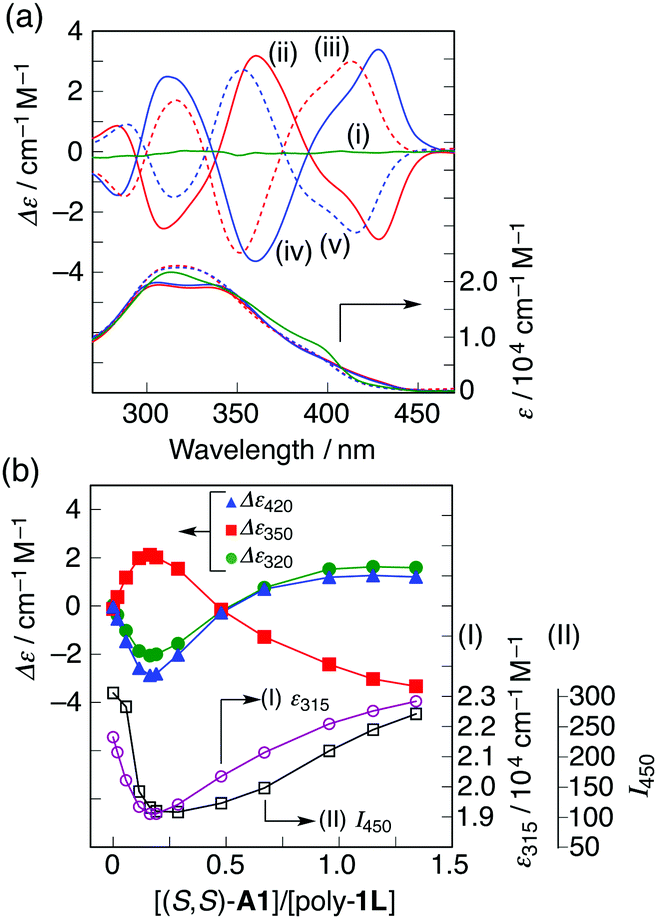 | ||
| Fig. 2 (a) CD and absorption spectra of poly-1L in the absence (i) and presence of (S,S)-A1 (ii, iii) and (R,R)-A1 (iv, v) in H2O/THF (6.5/3.5, v/v) at 25 °C: [poly-1L] = 6.8 × 10−5 M per monomer unit and [A1]/[poly-1L] = 0.19 (ii, iv) and 1.3 (iii, v). (b) Plots of CD intensity (left axis) at 420, 350 and 320 nm, molar absorptivity (right axis I) at 315 nm and fluorescence intensity (right axis II) at 450 nm of poly-1Lversus the molar ratio of (S,S)-A1 to poly-1L in H2O/THF (6.5/3.5, v/v) at 25 °C. | ||
The ICD intensity gradually increased with an increase in the amount of (S,S)-A1, along with both a significant reduction in the fluorescence intensity at around 450 nm and an apparent hypochromic effect in the absorption spectra, until an amount of A1 of 0.19 equiv. was reached (Fig. 2b and Fig. S3a, c, ESI†). Such a fluorescence quenching is typical for the helical folding of the PE-based foldamers due to a tight π–π stacking interaction between the non-adjacent PE units,1c,9 as supported by the close contact between the o-PE units (average distance of ca. 3.6 Å) in the energy-minimized structure of the left-handed helical poly-1 (9mer) (Fig. 3); the calculated CD spectrum matches well with the experimental one of poly-1L induced in the presence of a small amount of (S,S)-A1 (Fig. S4, ESI†). Therefore, the preferred-handed helical structure of poly-1L with the tight π–π stacking can be induced by complexation with the enantiopure A1, despite the resulting electrostatic repulsion between the negatively charged, spatially closed carboxylate groups at the side chains. This repulsive interaction might be compensated by the noncovalent intramolecular cross-linking of the two peripheral carboxylate groups along the helical poly-1L through the salt-bridge formation with two positively charged ammonium groups of A1 as a noncovalent cross-linker (Fig. 1b, (i)), thus leading to the close packing between the non-adjacent o-PE units, accompanied by the fluorescence quenching as already described. In sharp contrast, the fluorescence intensities at 415 and 450 nm of poly-1L were drastically enhanced by the increasing amount of monoamines (S)-A3 and (S)-A4 (Fig. S5 and S6, ESI†).9f,g This is likely due to a weak single electrostatic interaction between the ammonium and carboxylate groups of poly-1L that enhances the electrostatic repulsion between the carboxylate groups as a result of the dissociation of the ammonium–carboxylate ion pairs, thus leading to a random coil structure. Therefore, poly-1L showed no ICD in the presence of the chiral monoamines.
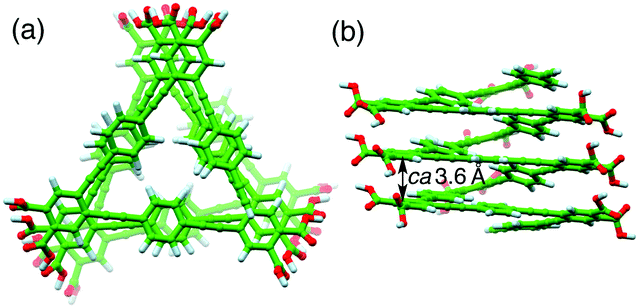 | ||
| Fig. 3 Top (a) and side (b) views of the energy-minimized left-handed helical structure of poly-1 (9mer) obtained via the semiempirical molecular orbital calculations. The average distance between two o-linked benzene rings at i and i + 3th positions is also shown. | ||
Interestingly, the further addition of (S,S)-A1 to the poly-1L complexed with 0.19 equiv. of (S,S)-A1 initially resulted in a significant decrease in the ICD intensity, followed by inversion of the Cotton effect signs concomitant with an apparent hyperchromic effect in the absorption spectra at higher concentrations of (S,S)-A1 (Fig. 2 and Fig. S3b, ESI†). This inversion of the Cotton effect signs was accompanied by a large increase in the fluorescence intensity at around 450 nm (Fig. S3d, ESI†), indicating that the poly-1L complexed with 1.3 equiv. of (S,S)-A1 possessed an opposite handed helical structure with a slightly loose stacking of the o-PE units as compared to the poly-1L with 0.19 equiv. of (S,S)-A1. The difference in the helical stacking structures of the o-PE units of poly-1L resulting from the different salt bridge formations (inter-residual and intra-residual, Fig. 1b) was further revealed by their thermal stability. As anticipated, the poly-1L complexed with 1.3 equiv. of (S,S)-A1 through intra-residual salt-bridges was highly sensitive to temperature and its ICD almost disappeared at 55 °C (Fig. 4b), whereas the ICD signal of the poly-1L with 0.19 equiv. of (S,S)-A1 was retained at 55 °C due to the noncovalent intramolecular cross-linking through the salt-bridge formation between the two peripheral carboxylate groups of the o-PE units and ammonium groups of A1 (Fig. 4a).
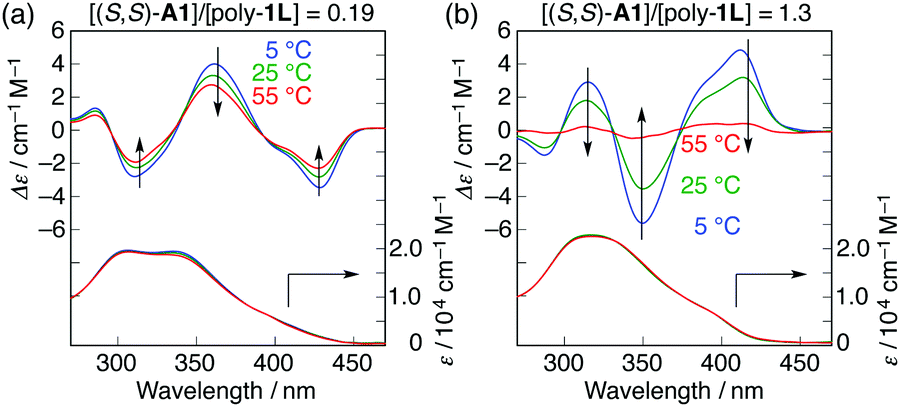 | ||
| Fig. 4 Temperature-dependent CD and absorption spectral changes of poly-1L in the presence of different amounts of (S,S)-A1 ([A1]/[poly-1L] = 0.19 (a) and 1.3 (b)) in H2O/THF (6.5/3.5, v/v): [poly-1L] = 6.8 × 10−5 M per monomer unit. | ||
When (S,S)-A2 was used instead of (S,S)-A1, a similar preferred-handed helicity induction of poly-1L was also observed, as evidenced by an ICD signal (Fig. S7a, ESI†). However, the complex did not exhibit inversion of the Cotton effects by the further addition of (S,S)-A2, although the absorption and fluorescence spectra changed similar to those observed for (S,S)-A1 (Fig. 2b and Fig. S7a–c, ESI†). The reason for this difference in the helix-inversion phenomena depending on the concentrations of (S,S)-A1 and (S,S)-A2 is not clear at present, but is considered to be due to the difference in their structural rigidity between A1 and A2. The linear diamine A2 is more flexible than the cyclic diamine A1, which may allow A2 to adjust its conformation to form inter-residual salt-bridges with the two spatially closed peripheral carboxylate groups of poly-1L independent of the concentration of A2 (Fig. 1b, (i)).13 In contrast, such an adjustable conformational change hardly occurred for the rigid cyclic diamine A1, resulting in a change to the intra-residual salt bridge formation at higher A1 concentrations (Fig. 1b, (ii)), thus leading to the helix-inversion.
Similar preferred-handed helicity induction and subsequent inversion of the helicity depending on the concentration of (S,S)- and (R,R)-A1 was also observed for the higher molecular weight poly-1H, and the complexes showed mirror image ICDs whose CD spectral patterns were similar to those of poly-1L (Fig. 5a and Fig. S8, ESI†), and (S,S)-A2 did not induce the helicity inversion of poly-1H (Fig. S9, ESI†). The maximum ICD intensities at 350 nm of poly-1H before and after the helicity inversion triggered by a change in the concentration of (S,S)-A1 were approximately 2.5 times greater than those of poly-1L (Fig. 2a and 5a). This is because the increasing main-chain length enhances the helical stability of the foldamers9b,e,g probably resulting from an inter-chain interaction at the side chains, which is also supported by the fact that the fluorescence spectrum of poly-1H alone was similar to that of the more closely packed helical poly-1L stabilized by 0.19 equiv. of (S,S)-A1 (Fig. S3c and S8c, ESI†). Moreover, the complex formation of poly-1H with the non-racemic A1 resulted in a positive nonlinear ICD response with respect to the enantiomeric excess (ee) of A1 (majority rule),14 indicating an amplification of the helix-sense excess that takes place during the folding process (Fig. 5b and Fig. S10a, ESI†), whereas the lower molecular weight poly-1L did not exhibit such a positive non-linearity (Fig. 5b and Fig. S11, ESI†).15 Interestingly, the ICD intensity of poly-1H complexed with 0.5 equiv. of the enantiopure and non-racemic A1 was significantly enhanced with time, resulting in a higher degree of the nonlinear ICD response (Fig. 5b, Fig. S10b and S12, ESI†). The observed higher amplification of the helical handedness excess of poly-1H may be ascribed to the aggregation of the induced helical poly-1H,16 whose size was roughly estimated to be ca. 20–100 nm by filtration experiments using membrane filters with different pore sizes (Fig. S13, ESI†).17 In sharp contrast, a mixture of poly-1L with 0.19 equiv. of (S,S)-A1 did not show such time-dependent ICD changes (Fig. S14, ESI†).
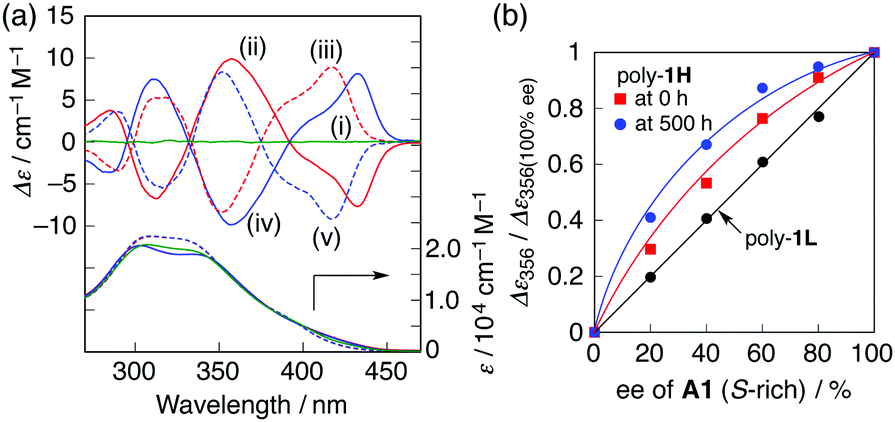 | ||
| Fig. 5 (a) CD and absorption spectra of poly-1H in the absence (i) and presence of (S,S)-A1 (ii, iii) and (R,R)-A1 (iv, v) measured immediately after the addition of A1 in H2O/THF (6.5/3.5 v/v) at 25 °C: [poly-1H] = 8.2 × 10−5 M per monomer unit and [A1]/[ poly-1H] = 0.2 (ii, iv) and 1.3 (iii, v). (b) Plots of normalized CD intensity at 356 nm of poly-1Hversus % ee value of A1 in H2O/THF (6.5/3.5, v/v) at 25 °C measured at 0 (red square) and 500 h (blue circle) after the addition of A1: [poly-1H] = 8.2 × 10−5 M per monomer unit and [(S,S)-A1]/[poly-1H] = 0.5. Plots of normalized CD intensity at 356 nm of poly-1Lversus % ee value of A1 are also shown (black circle): [poly-1L] = 6.8 × 10−5 M per monomer unit, [A1]/[poly-1L] = 0.19. | ||
In summary, we have demonstrated a preferred-handed helicity induction and subsequent inversion of the helicity triggered by the increasing amount of a single enantiomeric diamine in an optically inactive poly(o-PE-alt-p-PE)-based foldamer bearing carboxylic acid pendants, accompanied by a significant change in the stability of the helical structures. The detailed spectroscopic studies revealed that the stability difference between the pseudo-diastereomeric right- and left-handed helices is ascribed to the difference in the salt-bridge formations between the foldamer and the diamine that are determined by the concentration of the diamine. In addition, the longer foldamer showed an intriguing amplification of the helix-sense excess (the majority rule effect), which was further enhanced with time through supramolecular aggregation. The present results may provide a novel design strategy for the development of supramolecular asymmetric catalysts1g,19 and chiral materials for separating enantiomers20 based on the helicity-switchable foldamers.
This work was supported in part by JSPS KAKENHI (Grant-in-Aid for Scientific Research (S), no. 25220804 (E. Y.) and Grant Number JP16H01016 in Precisely Designed Catalysts with Customized Scaffolding (N. O.)). M. H. expresses her thanks for a JSPS Research Fellowship for Young Scientists (no. 10192).
Notes and references
- For recent reviews: (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS PubMed; (b) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS PubMed; (c) J. S. Moore and C. R. Ray, Adv. Polym. Sci., 2005, 177, 91 CrossRef; (d) Foldamers: Structure, Properties, and Applications, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (e) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed; (f) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed; (g) M. Suginome, T. Yamamoto, Y. Nagata, T. Yamada and Y. Akai, Pure Appl. Chem., 2012, 84, 1759 CrossRef CAS; (h) T. Nakano, Chem. Rec., 2014, 14, 369 CrossRef CAS PubMed; (i) J. Shen and Y. Okamoto, Chem. Rev., 2016, 116, 1094 CrossRef CAS PubMed; (j) E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33 RSC; (k) M. Fujiki, Symmetry, 2014, 6, 677 CrossRef CAS.
- (a) K. Maeda and Y. Okamoto, Macromolecules, 1998, 31, 5164 CrossRef CAS PubMed; (b) K. S. Cheon, J. V. Selinger and M. M. Green, Angew. Chem., Int. Ed., 2000, 39, 1482 CrossRef CAS; (c) M. Fujiki, J. Am. Chem. Soc., 2000, 122, 3336 CrossRef CAS; (d) K. Morino, K. Maeda and E. Yashima, Macromolecules, 2003, 36, 1480 CrossRef CAS; (e) K. Tang, M. M. Green, K. S. Cheon, J. V. Selinger and B. A. Garetz, J. Am. Chem. Soc., 2003, 125, 7313 CrossRef CAS PubMed; (f) J. Tabei, R. Nomura, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 1175 CrossRef CAS; (g) K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639 CrossRef CAS PubMed; (h) Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521 CrossRef CAS PubMed; (i) M. Shigeno, Y. Kushida and M. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 7972 CrossRef CAS PubMed.
- (a) K. Maeda, N. Kamiya and E. Yashima, Chem. – Eur. J., 2004, 10, 4000 CrossRef CAS PubMed; (b) T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899 CrossRef CAS PubMed; (c) Y. Nagata, T. Nishikawa and M. Suginome, J. Am. Chem. Soc., 2014, 136, 15901 CrossRef CAS PubMed.
- (a) S. Mayer, G. Maxein and R. Zentel, Macromolecules, 1998, 31, 8522 CrossRef CAS; (b) N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS PubMed; (c) J. Li, G. B. Schuster, K.-S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 2000, 122, 2603 CrossRef CAS; (d) D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693 CrossRef CAS PubMed; (e) Z. Yu and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 313 CrossRef CAS.
- (a) E. Yashima, Y. Maeda and Y. Okamoto, J. Am. Chem. Soc., 1998, 120, 8895 CrossRef CAS; (b) Y. Inai, Y. Ishida, K. Tagawa, A. Takasu and T. Hirabayashi, J. Am. Chem. Soc., 2002, 124, 2466 CrossRef CAS PubMed; (c) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed.
- (a) I. Otsuka, R. Sakai, T. Satoh, R. Kakuchi, H. Kaga and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5855 CrossRef CAS; (b) R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926 CrossRef CAS PubMed; (c) H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2008, 130, 792 CrossRef CAS PubMed; (d) S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868 CrossRef CAS PubMed; (e) F. Freire, J. M. Seco, E. Quinoa and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692 CrossRef CAS PubMed; (f) J.-m. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938 CrossRef CAS PubMed.
- (a) T. Miyagawa, A. Furuko, K. Maeda, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2005, 127, 5018 CrossRef CAS PubMed; (b) K. Maeda, T. Miyagawa, A. Furuko, H. Onouchi and E. Yashima, Macromolecules, 2015, 48, 4281 CrossRef CAS.
- (a) K. Maeda, K. Morino and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3625 CrossRef CAS; (b) T. Hasegawa, Y. Furusho, H. Katagiri and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 5885 CrossRef CAS PubMed; (c) M. Waki, H. Abe and M. Inouye, Angew. Chem., Int. Ed., 2007, 46, 3059 CrossRef CAS PubMed; (d) S. Takashima, H. Abe and M. Inouye, Chem. Commun., 2011, 47, 7455 RSC; (e) Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656 CrossRef CAS PubMed.
- (a) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793 CrossRef CAS PubMed; (b) R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114 CrossRef CAS; (c) T. V. Jones, M. M. Slutsky, R. Laos, T. F. A. de Greef and G. N. Tew, J. Am. Chem. Soc., 2005, 127, 17235 CrossRef CAS PubMed; (d) A. Khan and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1619 CrossRef CAS; (e) M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11 CrossRef CAS PubMed; (f) N. Zhu, W. Hu, S. Han, O. Wang and D. Zhao, Org. Lett., 2008, 10, 4283 CrossRef CAS PubMed; (g) N. Zhu, Q. Yan, Z. Luo, Y. Zhai and D. Zhao, Chem. – Asian J., 2012, 7, 2386 CrossRef CAS PubMed.
- An amide-bond formation between poly-1 and A1 could be ruled out based on a reaction between a model compound 7 (Fig. 1a) and 1.1 equiv. of (S,S)-A1 in D2O/THF-d8 (6.5/3.5, v/v) at 25 °C for 12 h.
- The CD spectrum of poly-2 in H2O/THF (6.5/3.5, v/v) was hardly changed upon the addition of 1.5 equiv. of (S,S)-A1 and (R,R)-A1.
- N. Ousaka, T. Yamaguchi and E. Yashima, Chem. Lett., 2014, 43, 512 CrossRef CAS.
- This hypothesis is supported by the fact that the ICD signal of poly-1L with 1.1 equiv. of (S,S)-A2 was retained at 55 °C (Fig. S7d, ESI†).
- (a) M. M. Green, B. A. Garetz, B. Munoz, H. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117, 4181 CrossRef CAS; (b) M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3139 CAS.
- For an example of molecular-weight-dependent enhancement of the majority rule effect, see: S. K. Jha, K.-S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 1999, 121, 1665 CrossRef CAS.
- For examples of aggregation-assisted amplification of helical chirality in the foldamers, see: (a) L. Brunsveld, E. W. Meijer, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 7978 CrossRef CAS PubMed; (b) W. Cai, G. T. Wang, P. Du, R. X. Wang, X. K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2008, 130, 13450 CrossRef CAS PubMed; (c) A. Petitjean, L. A. Cuccia, M. Schmutz and J. M. Lehn, J. Org. Chem., 2008, 73, 2481 CrossRef CAS PubMed; (d) Y. Wang, F. Li, Y. Han, F. Wang and H. Jiang, Chem. – Eur. J., 2009, 15, 9424 CrossRef CAS PubMed; (e) R. Pfukwa, P. H. J. Kouwer, A. E. Rowan and B. Klumperman, Angew. Chem., Int. Ed., 2013, 52, 11040 CrossRef CAS PubMed.
- Interestingly, the ICD signal of poly-1H retained for several hours upon the addition of a large excess (50 equiv.) of achiral ethylenediamine at 5 °C, suggesting a memory of the induced helicity.18 The further study for this phenomenon is currently underway in our laboratory.
- E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449 CrossRef CAS.
- (a) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660 RSC; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC; (c) A. Desmarchelier, X. Caumes, M. Raynal, A. Vidal-Ferran, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2016, 138, 4908 CrossRef CAS PubMed.
- K. Shimomura, T. Ikai, S. Kanoh, E. Yashima and K. Maeda, Nat. Chem., 2014, 6, 429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectroscopic data. See DOI: 10.1039/c6cc05753e |
| This journal is © The Royal Society of Chemistry 2016 |