
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of cyclic carbonates from diols and CO2 catalyzed by carbenes†
Felix D.
Bobbink‡
,
Weronika
Gruszka‡
,
Martin
Hulla
,
Shoubhik
Das
and
Paul J.
Dyson
*
Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
First published on 8th August 2016
Abstract
The synthesis of cyclic carbonates from epoxides and CO2 is a well-established reaction, whereas the synthesis of cyclic carbonates from diols and CO2 is considerably more challenging, and few efficient catalysts are available. Here, we describe heterocyclic carbene catalysts, including one derived from a cheap and efficient thiazolium salt, for this latter reaction. The reaction proceeds at atmospheric pressure in the presence of an alkyl halide and Cs2CO3. Reaction mechanisms for the transformations involved are also proposed.
Utilization of carbon dioxide (CO2) in the production of fine chemicals and synthetic fuels would contribute towards a more sustainable chemical industry. However, CO2 is a challenging molecule to activate as it is thermodynamically stable and kinetically inert in many transformations. Accordingly, only a few energy-efficient processes which employ CO2 have been commercialized.1
From a thermodynamic perspective, oxygenated cyclic carbonates are particularly suitable synthetic targets from CO2. These compounds have been exploited as electrolytes for lithium ion batteries,2 building blocks for polymeric materials,3,4 solvents5,6 and intermediates in the synthesis of compounds such as dimethyl carbonate (DMC)7 and ethylene glycol.8 Industrial production of cyclic carbonates involves either the transesterification of diols with phosgene in an energy-intensive process9 or the cycloaddition of CO2 to epoxides.10–12 Despite the latter route exhibiting 100% atom economy and industrial scalability, the synthesis of epoxides combined with their high reactivity and volatility are problematic. Recently, more stable, biodegradable 1,2-diols have been proposed as promising alternatives for the synthesis of cyclic carbonates with CO2.13 Their reaction with CO2 is, however, neither kinetically nor thermodynamically-favored due to the formation of water as the sole by-product.14 Attempts have been made to by-pass this problem by the implementation of a suitable catalyst system and a dehydrating agent. Both heterogeneous and homogeneous catalysts have been proposed for this reaction. For example, a heterogeneous cascade catalysis comprising CeO2 and 2-cyanopyridine is arguably the most efficient system.15 However, this process requires harsh reaction conditions (150 °C and 50 bars of CO2), an expensive reagent (2-cyanopyridine) and the activity is highly sensitive to the size of ceria particles. A number of homogeneous metal-free catalysts run under milder conditions and, interestingly, all are based on the 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) aided insertion of CO2. Different reagents are used to facilitate the subsequent alkylation step to afford cyclic carbonates in good yield under only 10 bars of CO2.16 The reaction may even proceed at an atmospheric pressure of CO2 if DBU and the alkyl halide are used in large excess.17 The same mild conditions are employed in a system in which tosyl chloride and triethylamine are used to afford cyclic carbonates with 6-membered rings in good yields.18 Ultimately, only a few efficient processes exist and finding an increasingly sustainable process for this reaction remains important.
Recently, N-heterocyclic carbenes (NHCs) have gained interest as catalysts for reactions which employ CO2 as a substrate.19–23 This stems from their ability to act as nucleophiles which activate CO2via the formation of imidazolium carboxylates.24,25 Interestingly, these intermediates have been previously reported to catalyze the synthesis of cyclic carbonates from diols employing DMC as the carbonyl source rather than CO2.26 Herein, we show the utility of carbene catalysts for the synthesis of cyclic carbonates from diols and CO2 and, based on key experiments, propose plausible mechanisms for this transformation.
Initially, reaction conditions were optimized using 1-phenyl-1,2-ethanediol (1a) as the substrate, see Table 1. Several imidazolium and thiazolium carbene catalysts (1c–4c) were evaluated. NHCs 1c and 3c19 and the thiazolium carbene catalysts 1b and 1d27 have been previously shown to catalyze the N-methylation of amines using CO2 as the carbon source. The efficiency of a variety of bases and alkyl halides was also studied as they are essential for the reaction to proceed (see below).16,18
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Alkyl halide (eq.) | Base (eq.) | Yield (%) |
| Reaction conditions: 1a (0.5 mmol), catalyst (20 mol%), alkyl halide (1–2.5 mmol), base (1–1.5 mmol), DMF (4 mL), CO2 (1 atm). Yields were determined by GC-FID using n-decane as internal standard.a The carbene catalyst was generated with NaH. Otherwise, the carbene is generated in situ using an extra 20 mol% base. | ||||
| 1 | 1c | CH2Br2 (2) | Cs2CO3 (2) | 44 |
| 2 | 2c | CH2Br2 (2) | Cs2CO3 (2) | 45 |
| 3 | 3c | CH2Br2 (2) | Cs2CO3 (2) | 29 |
| 4 | 4c | CH2Br2 (2) | Cs2CO3 (2) | 32 |
| 5 | 1c | CH2Br2 (5) | Cs2CO3 (2) | 42 |
| 6 | 2c | CH2Br2 (5) | Cs2CO3 (2) | 61 |
| 7 | 2c | CH2Br2 (5) | Cs2CO3 (3) | 53 |
| 8 | 2c | CH2Br2 (5) | Cs2CO3 (3) | 71 |
| 9 | 2c | CH2Br2 (5) | Na2CO3 (3) | 0 |
| 10 | 2c | CH2Br2 (5) | K2CO3 (3) | 5 |
| 11 | 2c | CH2Br2 (5) | DBU (3) | 21 |
| 12 | 2c | CH2Br2 (5) | Et3N (3) | 0 |
| 13 (50 °C) | 2c | CH2Br2 (5) | Cs2CO3 (3) | 5 |
| 14 (70 °C) | 2c | CH2Br2 (5) | Cs2CO3 (3) | 12 |
| 15 (110 °C) | 2c | CH2Br2 (5) | Cs2CO3 (3) | 25 |
| 16 | 2c | (CH2Br)2 (5) | Cs2CO3 (3) | 37 |
| 17 | 2c | (C2H4Br)2 (5) | Cs2CO3 (3) | 32 |
| 18 | 2c | C4H9Br (5) | Cs2CO3 (3) | 59 |
| 19 | 2c | C4H9Cl (5) | Cs2CO3 (3) | 20 |
| 20 | 2c | C4H9Br (2) | Cs2CO3 (3) | 81 |
| 21 (DMSO) | 2c | C4H9Br (2) | Cs2CO3 (3) | 33 |
| 22 (DMA) | 2c | C4H9Br (2) | Cs2CO3 (3) | 50 |
| 23 (Toluene) | 2c | C4H9Br (2) | Cs2CO3 (3) | 0 |
The ability of cesium carbonate (Cs2CO3) to activate CO2 and other small molecules28–32 encouraged us to employ it as a base in the reaction. Dibromomethane (CH2Br2) was also used due its efficiency in forming an effective leaving group.16 The activity of 1c–4c was investigated in the presence of 2 eq. of CH2Br2 and 2 eq. of Cs2CO3. The highest yields of styrene carbonate (1b) were obtained with catalysts 1c and 2c (Table 1, entries 1 and 2). In contrast, 3c and 4c resulted in lower product yields (Table 1, entries 3 and 4). The effect of quantities of CH2Br2 and Cs2CO3 on the reaction was studied. Increasing CH2Br2 to 5 eq. resulted in 61% yield of styrene carbonate 1b (Table 1, entry 6). Interestingly, a larger excess of the base (3 eq. instead of 2 eq.) led to a slight decrease in the yield of 1b (Table 1, entry 7). It should be noted that the reaction proceeds in low yield using Cs2CO3 as the base in the absence of CO2 (Table S2, entry 2, ESI†). However, 13C labeled CO2 was used to confirm that the main source of the carbonyl group incorporated in the cyclic carbonate product originates from CO2 (see NMR spectra comparing the 13C NMR spectra of non-labeled and 13C labeled products in the ESI†).
The enhanced activity of catalyst 2c might be due to a greater stability to moisture; note that 1b was not observed in a control experiment in which water was introduced into the system (Table S2, entry 6, ESI†). In the initial catalytic runs the active carbene catalyst was generated prior to reaction by the deprotonation of the corresponding salt with NaH. Subsequently, we found that the in situ generation of the carbene catalyst yielded 1b in 71% in presence of 3 eq. of Cs2CO3 (Table 1, entry 8). Interestingly, in a previous study using the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim][PF6]) as solvent, the increased carbonate yield was attributed to the increased solubility of CO2.16 Presumably, an active carbene was also generated by the deprotonation of the imidazolium salt by DBU – the ability of DBU and Cs2CO3 to deprotonate [bmim][BF4] to form a NHC has been reported.33 DBU was evaluated under our conditions, but yielded 1b in a significantly lower yield (Table 1, entry 11).16 Na2CO3 and K2CO3 were evaluated in place of Cs2CO3, but afford the product in 0 and 5% yield, respectively, presumably due to the lower solubility of these carbonates in DMF (Table 1, entries 9 and 10). No product was observed with Et3N (Table 1, entry 12). We speculate that Et3N, which is often employed in the Stetter reaction, may undergo a Menshutkin reaction with CH2Br2 thereby inhibiting the reaction. Notably, Cs2CO3 was found to be the optimal base in this reaction owing to its ability to generate the active carbene catalyst as well as to act as a minor carbonyl donor and a dehydrating agent.
Dimethylformamide (DMF) was selected as a reaction solvent as it can activate CO2.34 As expected, other polar aprotic solvents (dimethyl sulfoxide (DMSO) or dimethylacetamide (DMA)) could also be used (Table 1, entries 21 and 22), whereas no reaction was observed in toluene (Table 1, entry 23).
The optimum reaction temperature is 90 °C, with lower temperatures leading to a decrease in product yield (Table 1, entries 13 and 14) and with more elevated temperatures, e.g. 110 °C, leading to deactivation of the catalytic system (Table 1, entry 15). The alkyl halide also affects the reaction, in particular, 2 eq. of bromobutane (C4H9Br) results in a higher yield than 5 eq. of CH2Br2 (Table 1, entries 8 and 20). The other alkyl halides evaluated were less effective (Table 1, entries 16, 17 and 19).
Based on the optimized conditions, which afford 1b in up to 81% yield, the scope of the reaction was explored using catalyst 2c (Table 2). The substrates varied from 1,2-diols to 1,3-diols (2a–4a) bearing functional groups with varying steric influence. The diols were subjected to the optimized conditions of 2 eq. bromobutane, 3.2 eq. Cs2CO3 at 90 °C and 1 atm CO2 pressure.
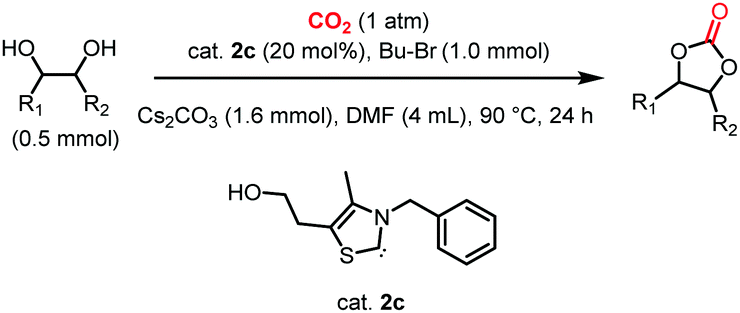
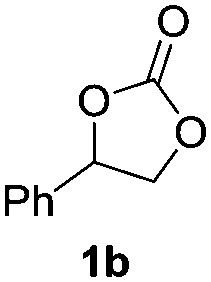
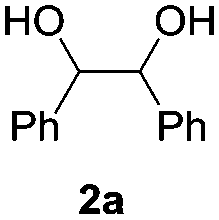
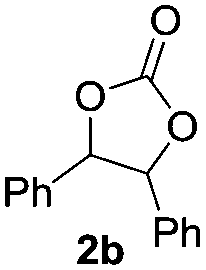
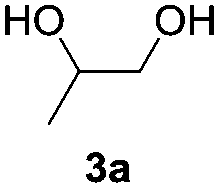
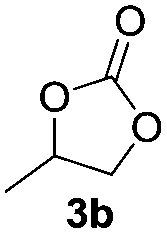
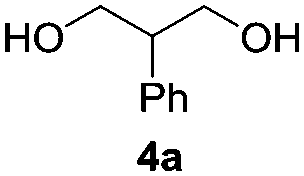
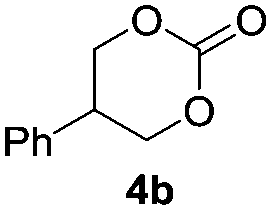
The model product 4-phenyl-1,3-dioxolan-2-one was isolated in 61% yield (Table 2, entry 1). Five-membered cyclic carbonates, 4,5-diphenyl-1,3-dioxolan-2-one (2b) and propylene carbonate (3b) were obtained in yields of 63 and 54%, respectively (Table 2, entries 2 and 3). The six-membered cyclic carbonate, 5-phenyl-1,3-dioxan-2-one (4b) was produced in 53% (Table 2, entry 4). These yields are comparable to those obtained using alternative methods (see Table S1, ESI† for a comparison).16,17
On the basis of our results and previous literature, two plausible reaction mechanisms in Schemes 1 and 2 are suggested.16,26,35,36Scheme 1 presents the principal mechanism for the carbene-catalyzed reaction. As mentioned above, both the base and alkyl halide are essential in the reaction, as confirmed in control experiments in which no carbonate was formed in their absence (Table S1, entries 4 and 5, ESI†). Scheme 2 represents the mechanism for the minor non-catalytic formation of cyclic carbonate in the absence of CO2 (Table S1, entry 2, ESI†). C4H9Br is included in the second mechanism due to detection of dibutyl carbonate and n-butanol in the reaction mixture using GC-MS, see ESI.† However, a similar mechanism is likely to take place in presence of other alkyl halides. Moreover, both of these mechanisms appear to occur concurrently to form the cyclic carbonate. This hypothesis is based on our finding that while 25% of 1b was obtained in the absence of CO2 (Table S1, entry 2, ESI†), addition of CO2 increased the yield of 1b to 81% (Table 1, entry 17).
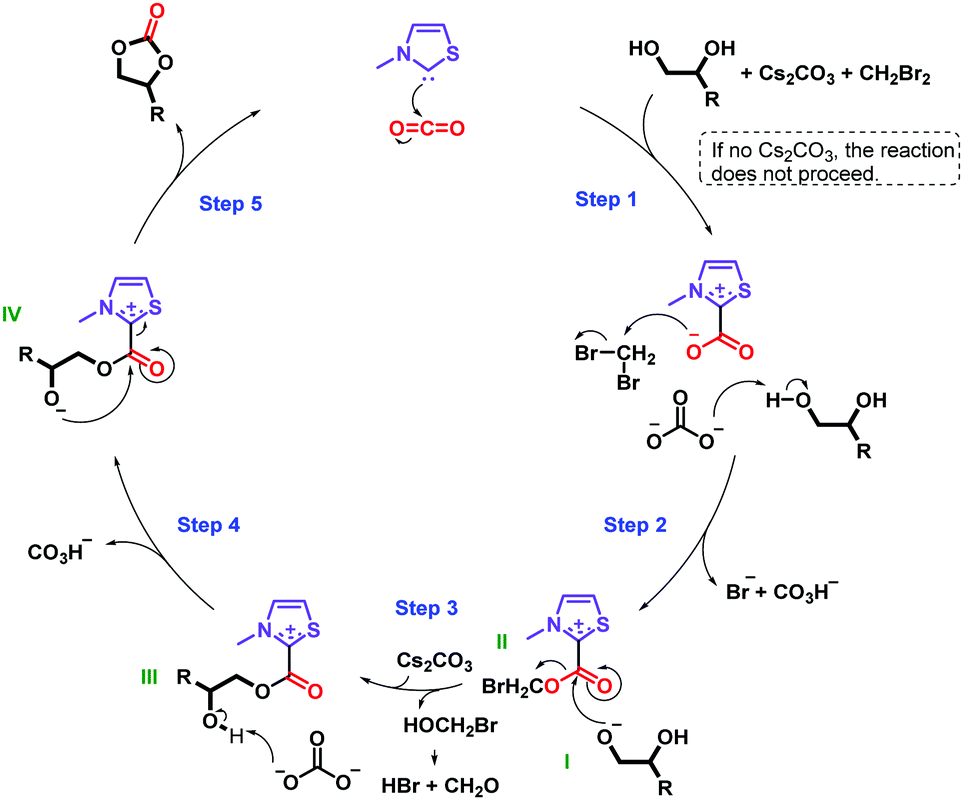 | ||
| Scheme 1 Tentative mechanism for the carbene-catalyzed reaction of diols and CO2 to form cyclic carbonates. The substituents of the catalyst are omitted for clarity. | ||
 | ||
| Scheme 2 Proposed mechanism for the non-catalytic reaction of diols and CO2 to form cyclic carbonates. | ||
In the mechanism in Scheme 1, step 2 involves the generation of an alkoxide I and the parallel attack of the carbene–CO2 adduct on CH2Br2 after activation of CO2 by the carbene in step 1. Nucleophilic attack of the alkoxide I on intermediate II in step 3 results in the elimination of the leaving group and formation of intermediate III. In step 4, the secondary hydroxyl group of the diol is deprotonated, leading to the generation of intermediate IV and, in the final step (step 5), the intramolecular addition of the alcohol occurs in intermediate IV, which affords the cyclic carbonate and regenerates the catalyst. Notably, bromomethanol is eliminated as a leaving group, however bromomethanol is unstable, and hence it is believed to decompose to a mixture of hydrogen bromide (HBr) and formaldehyde (CH2O).37 Note, the formation of these side-products was not detected by spectroscopic or chromatographic studies, possibly due to neutralization of HBr by Cs2CO3 and the volatility of CH2O.
The secondary (non-catalytic) reaction in Scheme 2 proceeds by the attack of Cs2CO3 on C4H9Br in step 1, leading to the formation of intermediate II (dibutyl carbonate was observed by GC-MS). Similar to the mechanism in Scheme 1, the reaction of the alkoxide I with intermediate II in step 2 leads to the elimination of butanol (observed by GC-MS) and the formation of intermediate III. Again, the deprotonation of the secondary hydroxyl group in intermediate III in step 3 results in the formation of intermediate IV. The final cyclization in step 4 leads to the elimination of the second leaving group and the formation of the cyclic carbonate.
In summary, the work presented here offers an approach for the synthesis of cyclic carbonates from diols and CO2. The proposed system benefits from the use of environmentally-friendly metal-free carbene catalysts. Using this methodology cyclic carbonates were obtained under mild conditions (90 °C and atmospheric pressure of CO2) in good yield and comparable or better to those obtained with other catalysts that operate under more forcing conditions. Based on labelling studies and other experiments two-mechanisms are proposed, one non-catalytic and one catalytic that account for the overall reaction.
The EPFL, the CTI Swiss Competence Center of Energy Research (SCCER) on Heat and Electricity Storage and the Fondation Claude et Juliana are thanked for funding.
References
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 1–15 Search PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- B. Nohra, L. Candy, C. Guerin and Y. Raoul, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- W. Guerin, A. K. Diallo, E. Kirilov, M. Helou, M. Slawinski, J. M. Brusson, J. F. Carpentier and S. M. Guillaume, Macromolecules, 2014, 47, 4230–4235 CrossRef CAS.
- C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420–3432 CrossRef CAS PubMed.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- P. Wang, S. Liu, F. Zhou, B. Yang, A. S. Alshammari, L. Lu and Y. Deng, Fuel Process. Technol., 2014, 126, 359–365 CrossRef CAS.
- C. Lian, F. Ren, Y. Liu, G. Zhao, Y. Ji, H. Rong, W. Jia, L. Ma, H. Lu, D. Wang and Y. Li, Chem. Commun., 2015, 51, 1252–1254 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584 RSC.
- F. D. Bobbink and P. J. Dyson, J. Catal., 2016 DOI:10.1016/j.jcat.2016.02.033.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- K. Müller, L. Mokrushina and W. Arlt, Chem. Ing. Tech., 2014, 86, 497–503 CrossRef.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 4824–4831 Search PubMed.
- Y. N. Lim, C. Lee and H. Y. Jang, Eur. J. Org. Chem., 2014, 1823–1826 CrossRef CAS.
- T. Kitamura, Y. Inoue, T. Maeda and J. Oyamada, Synth. Commun., 2016, 46, 39–45 CrossRef CAS.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- S. Das, F. D. Bobbink, G. Laurenczy and P. J. Dyson, Angew. Chem., Int. Ed., 2014, 53, 12876–12879 CrossRef CAS PubMed.
- O. Jacquet, C. Das Neves Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- S. Das, F. D. Bobbink, A. Gopakumar and P. J. Dyson, Chimia, 2015, 69, 765–768 CrossRef CAS PubMed.
- A. M. Voutchkova, M. Feliz, E. Clot, O. Eisenstein and R. H. Crabtree, J. Am. Chem. Soc., 2007, 129, 12834–12846 CrossRef CAS PubMed.
- S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed.
- J. A. Stewart, R. Drexel, B. Arstad, E. Reubsaet, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2016, 18, 1605–1618 RSC.
- S. Das, F. D. Bobbink, S. Bulut, M. Soudani and P. J. Dyson, Chem. Commun., 2016, 52, 2497–2500 RSC.
- A. Ion, V. Parvulescu, P. Jacobs and D. De Vos, Green Chem., 2007, 9, 158–161 RSC.
- R. N. Salvatore, S. I. Shin, A. S. Nagle and K. W. Jung, J. Org. Chem., 2001, 66, 1035–1037 CrossRef CAS PubMed.
- Y. P. Patil, P. J. Tambade, S. R. Jagtap and B. M. Bhanage, Green Chem. Lett. Rev., 2008, 1, 127–132 CrossRef CAS.
- T. Niemi, J. E. Perea-Buceta, I. Fernandez, O.-M. Hiltunen, V. Salo, S. Rautiainen, M. T. Räisänen and T. J. Repo, Chem. – Eur. J., 2016, 22, 1–6 CrossRef PubMed.
- D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1–6 CrossRef PubMed.
- I. Chiarotto, M. Feroci, G. Forte, M. Orsini and A. Inesi, ChemElectroChem, 2014, 1, 1525–1530 CrossRef CAS.
- B. Liu, A. G. Wong-Foy and A. J. Matzger, Chem. Commun., 2013, 49, 1419–1421 RSC.
- A. Schmidt, A. Beutler, M. Albrecht, B. Snovydovych and F. J. Ramírez, Org. Biomol. Chem., 2008, 6, 287–295 CAS.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- A. R. Katritzky, et al., Comprehensive Organic Functional Group Transformations: Synthesis: carbon with one heteroatom attached by a single bond, Elsevier, 1995, vol. 2 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental and spectroscopy data. See DOI: 10.1039/c6cc05730f |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |