
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activation of SO2 by [Zn(Cp*)2] and [(Cp*)ZnI–ZnI(Cp*)]†
Rory P.
Kelly
a,
Neda
Kazeminejad
a,
Carlos A.
Lamsfus
b,
Laurent
Maron
b and
Peter W.
Roesky
*a
aInstitute of Inorganic Chemistry Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu; Fax: +49 721 6084 4854
bLPCNO, CNRS & INSA, Université Paul Sabatier, 135 Avenue de Rangueil, Toulouse 31077, France
First published on 26th September 2016
Abstract
Interesting reactivity was observed in reactions of SO2 with [Zn(Cp*)2] and [(Cp*)ZnI–ZnI(Cp*)]. These reactions proceeded with insertion of SO2 into the Zn–C bonds. Spectacularly, the lability of the C–S bond in the O2SCp* ligands led to the thermal decomposition of [Zn(O2SCp*)2(tmeda)] to afford [Zn2(μ-SO3)(μ-S2O4)(tmeda)2].
The reduction of SO2 to elemental sulfur (the Claus process) or sodium dithionite are important industrial reactions, but the primary use of SO2 is in the manufacture of sulfuric acid via the contact process. Befitting its industrial importance, there has been renewed academic interest in the activation of SO2 by molecular species spanning much of the periodic table. Reactions of low-valence complexes with SO2 can form dithionite complexes1 but insertion of SO2 into M–O2 or M–C bonds,1b,c,3 affording sulfite and sulfinate complexes, respectively, is also known, along with other reactions.4 However, despite these recent advances, reactions of SO2 with zinc complexes remain poorly studied, with the few reports thus far only detailing insertion reactions.3,5 Herein, we report the reactions between SO2 and the zinc complexes, [Zn(Cp*)2] and [(Cp*)ZnI–ZnI(Cp*)] (Cp* = C5Me5−).
When two equivalents of SO2 were condensed onto a stirring solution of [Zn(Cp*)2] in thf, a white solid precipitated immediately. The white solid that formed did not dissolve to any appreciable extent, even with heating. However, when the same reaction was performed in the presence of tmeda (tmeda = N,N′-tetramethylethylenediamine), a clear solution was obtained. From this solution, the SO2 insertion product, [Zn(O2SCp*)2(tmeda)] (1), was obtained in good yield (Scheme 1).
 | ||
| Scheme 1 Syntheses of [Zn(O2SCp*)2(tmeda)] (1) and [Zn4(O2SCp*)6O] (2). | ||
Complex 1 crystallised from thf in the monoclinic space group, P2/c. The molecular structure and selected bond lengths are listed in Fig. 1. The six-coordinate zinc atom features a distorted octahedral coordination geometry environment, reminiscent of related carboxylate complexes, e.g. [Zn{O2C(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)CH3}2(tmeda)]6 and [Zn(O2CCH3)2(tmeda)].7 The Zn–O bond lengths (2.1139(12) and 2.2340(12) Å) are unequal in length, but this is also observed in the aforementioned carboxylate complexes, and it is presumably a result of the hexacoordination of the zinc atom in complex 1. It is a rare example of a Zn sulfinate complex, although other examples have been reported.3,5
CH)CH3}2(tmeda)]6 and [Zn(O2CCH3)2(tmeda)].7 The Zn–O bond lengths (2.1139(12) and 2.2340(12) Å) are unequal in length, but this is also observed in the aforementioned carboxylate complexes, and it is presumably a result of the hexacoordination of the zinc atom in complex 1. It is a rare example of a Zn sulfinate complex, although other examples have been reported.3,5
 | ||
| Fig. 1 Molecular structure of [Zn(O2SCp*)2(tmeda)] (1). Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Zn(1)–O(1) = 2.1139(12), Zn(1)–O(2) = 2.2340(12), Zn(1)–N(1) = 2.1665(14). Half of the molecule is generated by symmetry. | ||
When [(Cp*)ZnI–ZnI(Cp*)] was treated with SO2, complete oxidation of ZnI to ZnII was observed, and the zinc oxo-cluster, [Zn4(O2SCp*)6O] (2), was obtained in moderate yield (Scheme 1). This reaction was performed multiple times and 2 is the only product that has been isolated to date. Complex 2 evidently arose from insertion of SO2 into the Zn–Cp* bonds, analogously to 1, and possibly reduction of SO2 or an in situ formed derivative by the Zn–Zn bond to abstract an oxygen atom, but other pathways cannot be ruled out.8 We do not think that water is a likely source of the oxide ion since we used high purity SO2 and complex 2 was isolated multiple times from different batches of [(Cp*)ZnI–ZnI(Cp*)].9 Furthermore, we did not ever observe the formation of Zn metal, which would accompany disproportionation, and thus we believe that the Zn–Zn unit is oxidised by SO2 or an in situ formed derivative, e.g. the O2SCp* ligands. It is difficult to rule out other sources, e.g. thf cleavage, and attempts to do so have so far been inconclusive. Nonetheless, our case quite possibly represents a rare case of deoxygenation of SO2 by a low-valent metal complex. The reductive cleavage of SO2 into SO and O2− is generally unfavourable due to the instability of SO, and this highlights the impressive reducing ability of ZnI in the current case, when even trivalent uranium2b or divalent lanthanide1b,c systems, which feature highly reducing and oxophilic metal centres, have proven incapable of abstracting an oxygen atom from SO2. It is worth noting that it has been reported that CO2 does not react with [(Cp*)ZnI–ZnI(Cp*)].10 We repeated this reaction and found the same result.
Complex 2·1.5(C5H12) crystallised from pentane in the triclinic space group, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The complex has a pentanuclear [Zn4O] core featuring four zinc atoms coordinated tetrahedrally around a central oxide ion. Each Zn atom is further ligated by one oxygen atom of three separate O2SCp* ligands, with six such ligands in total. Such a [Zn4O] core is a common feature in zinc-oxo complexes, and it is also a very popular node in MOF chemistry.11 The zinc atoms are all four coordinate, in contrast to the six-coordinate zinc atom in complex 1. The distorted tetrahedral coordination sphere of each zinc atom is completed by coordination to one oxygen atom of three separate bridging O2SCp* ligands, giving six such ligands in the complex (Fig. 2).
. The complex has a pentanuclear [Zn4O] core featuring four zinc atoms coordinated tetrahedrally around a central oxide ion. Each Zn atom is further ligated by one oxygen atom of three separate O2SCp* ligands, with six such ligands in total. Such a [Zn4O] core is a common feature in zinc-oxo complexes, and it is also a very popular node in MOF chemistry.11 The zinc atoms are all four coordinate, in contrast to the six-coordinate zinc atom in complex 1. The distorted tetrahedral coordination sphere of each zinc atom is completed by coordination to one oxygen atom of three separate bridging O2SCp* ligands, giving six such ligands in the complex (Fig. 2).
 | ||
| Fig. 2 Molecular structure of [Zn4(O2SCp*)6O]·1.5(C5H12) (2·1.5(C5H12)). Hydrogen atoms and lattice solvent have been omitted for clarity. One of the O2SCp* moieties (corresponding to S(6)) is disordered over two positions, so only one of the conformations is shown. Selected bond lengths (Å): Zn(1)–O(1) = 1.939(5), Zn(1)–O(6) = 1.943(5), Zn(1)–O(9) = 1.969(5), Zn(1)–O(13) = 1.958(6). | ||
Complex 2 is the sulfinate analogue of [Zn4(O2CCp*)6O].11c In contrast to complex 1, the Zn–O(sulfinate) bond lengths are shorter (1.90(2)–1.99(2) Å), and this is presumably a function of the lower coordination number in complex 2. The lower coordination in complex 2 also accounts for the shorter Zn–O bond lengths. The Zn–O(oxide) bond lengths also fall in the same range as the Zn–O(sulfinate) bond lengths. Unsurprisingly, the Zn–O bond lengths in 2 are very similar to those in [Zn4(O2CCp*)6O].
Both 1 and 2 were characterised by NMR (vide infra) and IR spectroscopy, and satisfactory microanalyses were obtained for both complexes.
1H NMR studies of 1 and 2 revealed that the complexes are thermally unstable and that they slowly decompose in solution to form multiple products. An interesting feature of 1 is that the O2SCp* ligands only give rise to two broad signals, in contrast to the three sharp singlets in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio observed for O2CCp*11c or S2CCp*12 ligands bound to Zn cations. The broadness of the signals for the O2SCp* ligands is indicative of rapid exchange of the Cp* ring carbon atoms and the sulfur atom of the SO2 moiety. This hinted that the instability of the complexes was due to the lability of the C–S bond in the O2SCp* ligands. The 1H NMR spectrum of complex 2 is much more complex than that of 1, and no obvious assignment has yet proved possible, even after obtaining spectra at low temperatures. It seems likely that there is dissociation of the complex into multiple species upon dissolution.
:2:1 ratio observed for O2CCp*11c or S2CCp*12 ligands bound to Zn cations. The broadness of the signals for the O2SCp* ligands is indicative of rapid exchange of the Cp* ring carbon atoms and the sulfur atom of the SO2 moiety. This hinted that the instability of the complexes was due to the lability of the C–S bond in the O2SCp* ligands. The 1H NMR spectrum of complex 2 is much more complex than that of 1, and no obvious assignment has yet proved possible, even after obtaining spectra at low temperatures. It seems likely that there is dissociation of the complex into multiple species upon dissolution.
Much to our surprise, monitoring the decomposition of 1 by 1H NMR spectroscopy showed that one of the decomposition products was (C5Me5)2. The origin of this species is the coupling of two Cp*˙ radicals, which form from the loss of one electron per Cp* anion (eqn (1)). Evans has demonstrated that [Ln(Cp*)3] complexes are highly reducing despite having the lanthanide ions in their highest accessible oxidation state, and the area has been termed sterically-induced reduction.13 Similar Cp*-based reductions have recently been observed in Zn chemistry,14 but base-induced reduction from a functionalised Cp* ligand is, to the best of our knowledge, unprecedented.
| 2(C5Me5)− → (C5Me5)2 + 2e− | (1) |
In an effort to study these intriguing decomposition reactions, complex 1 was generated in situ and then heated at 70 °C for several hours. Although the decomposition reaction is complex, we isolated single crystals of [Zn2(μ-SO3)(μ-S2O4)(tmeda)2] (3) in modest yield (Scheme 2). Importantly, the reaction is repeatable. Complex 3 is highly insoluble, thus preventing its characterisation by NMR spectroscopy, but it was characterised by X-ray crystallography, IR spectroscopy and microanalysis.
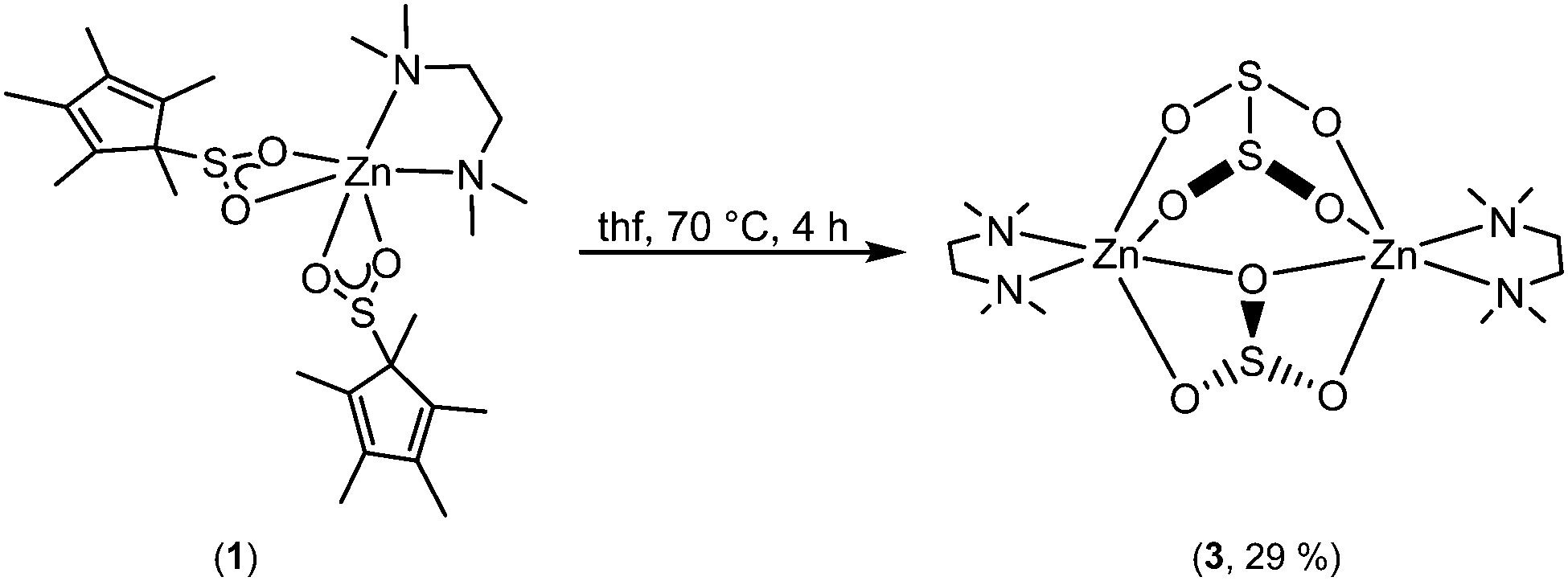 | ||
| Scheme 2 Formation of [Zn2(μ-SO3)(μ-S2O4)(tmeda)2] (3) from the thermal decomposition of [Zn(O2SCp*)2(tmeda)] (1). | ||
Complex 3·thf crystallised from thf in the monoclinic space group, P21/c. The molecular structure is shown in Fig. 3, along with selected bond lengths. The dinuclear complex features two six-coordinate zinc atoms that are each bound to one tmeda ligand, one bridging dithionite ligand and one bridging sulfite ligand. The binding of the sulfite and dithionite ligands to the two zinc atoms is slightly asymmetric. Surprisingly, to the best of our knowledge the only other Zn complex containing dithionite ligands is the simple Zn salt, [Zn(S2O4)(NC5H5)].15 On the other hand, there are multiple examples of Zn sulfite complexes, but they remain a fairly limited class, and most examples feature polymeric arrays. However, related bimetallic complexes such as [Zn(μ-SO3)(phen)]216 (phen = 1,10-phenanthroline) and [Zn(μ-SO3)(bipy)]217 (bipy = 2,2′-bipyridine) are known.
 | ||
| Fig. 3 Molecular structure of [Zn2(μ-SO3)(μ-S2O4)(tmeda)2]·thf (3·thf). Hydrogen atoms and lattice solvent have been omitted for clarity. Selected bond lengths (Å): Zn(1)–O(1) = 2.142(2), Zn(1)–O(3) = 2.162(2), Zn(1)–O(4) = 2.100(2), Zn(1)–O(6) = 2.143(2), Zn(1)–N(1) = 2.203(3), Zn(2)–O(1) = 2.096(2), Zn(2)–O(2) = 2.347(3), Zn(2)–O(5) = 2.103(2), Zn(1)–O(7) = 2.099 (2), Zn(2)–N(3) = 2.189(3). | ||
Whilst it is difficult to state exactly what occurs during this decomposition reaction, the presence of the sulfite ligand (SO32−) can be rationalised by a combination of deoxygenation of an SO2 moiety to yield a Zn oxo species, and subsequent insertion of another molecule of SO2 into the Zn–O bond to form the sulfite ligand. Recently, we showed that SO2 can insert into a metal oxide bridge (M–O–M) to give the corresponding sulfite complex, (M–SO3–M).2a The dithionite ligand (S2O42−) presumably arises from homolytic cleavage of the C–S bond in O2SCp*, formally giving the two radical species, SO2˙− and Cp*˙. Both of these species can couple to form S2O42− and (C5Me5)2, respectively, the latter of which was detected in solution. Subtle changes in reaction conditions presumably affect the outcome of these reactions but it is a spectacular demonstration of the reactivity of these complexes. The decomposition of 1 to afford 3 was deemed to be too complicated to model computationally but we modelled the insertion of SO2 into [Zn(Cp*)2] to yield 1 (Fig. 4).
 | ||
| Fig. 4 Computed enthalpy profile (kcal mol−1) for the reaction of SO2 with [Zn(Cp*)2] at room temperature. | ||
The reaction begins with the coordination of a tmeda molecule to the zinc atom, inducing a haptotropic shift of the originally η5-Cp* ligand, which then becomes sigma-bonded through one carbon atom. This shift was found to be endothermic by 10.1 kcal mol−1 but it is crucial for the subsequent reactivity with SO2. Indeed, from this complex, the approach of SO2 leads to the partial decoordination of a Cp* ligand, which is then able to nucleophilically attack the incoming SO2 molecule. Starting from the tmeda-coordinated complex, the barrier for such an attack is very low (4.9 kcal mol−1 or 15.0 kcal mol−1 with respect to the tmeda-free decamethylzinconcene). This reaction yields an intermediate that appears to be quite stable (−33.7 kcal mol−1 with respect to the entrance channel). In this intermediate, the inserted SO2 molecule only interacts with the Zn centre through one oxygen atom in order to maintain the four-fold coordination around the metal. The approach of a second SO2 molecule leads to a similar process as described before. Indeed, the second Cp* ligand gets decoordinated and nucleophilically attacks the coordinated SO2. The barrier for this second insertion is negligible (a few tenths of a kcal mol−1), in line with a facile process. As for the first insertion, this second one is strongly exothermic by 29.7 kcal mol−1, yielding the formation of complex 1. The Cp* ligand, often considered an innocent ligand, reacts like an alkyl group. Such reactivity appears to be enhanced by the sigma-coordination of the Cp* ligand in the complex (favoured by the coordination of tmeda), and this has been observed before in zinc Cp* chemistry.11c,12,14b
In summary, the reactions of the zinc complexes, [Zn(Cp*)2] and [(Cp*)ZnI–ZnI(Cp*)], with SO2 yielded the sulfinate complex, [Zn(O2SCp*)2(tmeda)] (1), and the sulfinate/oxo cluster, [Zn4(O2SCp*)6O] (2), respectively. In both cases, nucleophilic attack of the Cp* ligand on the incoming SO2 molecule was observed, and in the reaction with [(Cp*)ZnI–ZnI(Cp*)], complete oxidation of the ZnI–ZnI bond was observed. DFT calculations clearly show the decoordination of the Cp* ligand, followed by nucleophilic attack on the SO2 molecule, and finally, coordination of the newly formed O2SCp*− sulfinate anion. Somewhat surprisingly, these complexes are thermally unstable due to the lability of the C–S bond of the sulfinate ligands. In the case of complex 1, thermally-induced decomposition allowed us to isolate the mixed dithionite/sulfite complex, [Zn2(μ-SO3)(μ-S2O4)(tmeda)2] (3), in modest yield. This multi-step activation of SO2 is noteworthy since activation of SO2 by molecular systems tends to stop after insertion or coupling.
This work was supported by the DFG-funded transregional collaborative research center SFB/TRR 88 “3MET”. LM would like to thank the Alexander-von-Humboldt Foundation, Chinese Academy of Science and ANR for financial support. CalMip is also gratefully acknowledged for a generous grant of computing time.
Notes and references
- (a) K. Junold, F. M. Mück, C. Kupper, J. A. Baus, C. Burschka and R. Tacke, Chem. – Eur. J., 2014, 20, 12781–12785 CrossRef CAS PubMed; (b) S. V. Klementyeva, N. Arleth, K. Meyer, S. N. Konchenko and P. W. Roesky, New J. Chem., 2015, 39, 7589–7594 RSC; (c) S. V. Klementyeva, M. T. Gamer, A.-C. Schmidt, K. Meyer, S. N. Konchenko and P. W. Roesky, Chem. – Eur. J., 2014, 20, 13497–13500 CrossRef CAS PubMed; (d) A. J. Boutland, I. Pernik, A. Stasch and C. Jones, Chem. – Eur. J., 2015, 21, 15749–15758 CrossRef CAS PubMed; (e) A. Noor, S. Qayyum, T. Bauer, S. Schwarz, B. Weber and R. Kempe, Chem. Commun., 2014, 50, 13127–13130 RSC.
- (a) C. Schoo, S. V. Klementyeva, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2016, 52, 6654–6657 RSC; (b) A.-C. Schmidt, F. W. Heinemann, C. E. Kefalidis, L. Maron, P. W. Roesky and K. Meyer, Chem. – Eur. J., 2014, 20, 13501–13506 CrossRef CAS PubMed.
- (a) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S.-i. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed; (b) R. Eberhardt, M. Allmendinger, G. A. Luinstra and B. Rieger, Organometallics, 2003, 22, 211–214 CrossRef CAS.
- (a) L. Gonzalez-Sebastian, M. Flores-Alamo and J. J. Garcia, Dalton Trans., 2011, 40, 9116–9122 RSC; (b) P. Benndorf, S. Schmitt, R. Köppe, P. Oña-Burgos, A. Scheurer, K. Meyer and P. W. Roesky, Angew. Chem., Int. Ed., 2012, 51, 5006–5010 CrossRef CAS PubMed; (c) M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- M. F. Pilz, C. Limberg, B. B. Lazarov, K. C. Hultzsch and B. Ziemer, Organometallics, 2007, 26, 3668–3676 CrossRef CAS.
- D. A. Brown, N. J. Fitzpatrick, H. Müller-Bunz and Á. T. Ryan, Inorg. Chem., 2006, 45, 4497–4507 CrossRef CAS PubMed.
- D. A. Brown, W. Errington, N. J. Fitzpatrick, W. K. Glass, T. J. Kemp, H. Nimir and A. T. Ryan, Chem. Commun., 2002, 1210–1211 RSC.
- (a) M. A. Chilleck, T. Braun and B. Braun, Chem. – Eur. J., 2011, 17, 12902–12905 CrossRef CAS PubMed; (b) S. Mebs, M. A. Chilleck, S. Grabowsky and T. Braun, Chem. – Eur. J., 2012, 18, 11647–11661 CrossRef CAS PubMed.
- I. Resa, E. Carmona, E. Gutierrez-Puebla and A. Monge, Science, 2004, 305, 1136 CrossRef CAS PubMed.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693–703 CrossRef CAS PubMed.
- (a) K. Zelga, M. Leszczynski, I. Justyniak, A. Kornowicz, M. Cabaj, A. E. H. Wheatley and J. Lewinski, Dalton Trans., 2012, 41, 5934–5938 RSC; (b) T. Cadenbach, J. R. Pankhurst, T. A. Hofmann, M. Curcio, P. L. Arnold and J. B. Love, Organometallics, 2015, 34, 2608–2613 CrossRef CAS; (c) S. Schulz, S. Schmidt, D. Bläser and C. Wölper, Eur. J. Inorg. Chem., 2011, 4157–4160 CrossRef CAS; (d) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed; (e) A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494–3495 CrossRef CAS PubMed.
- S. Schulz, S. Schmidt, D. Bläser and C. Wölper, Z. Anorg. Allg. Chem., 2012, 638, 1705–1710 CrossRef CAS.
- (a) W. J. Evans, K. J. Forrestal and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 9273–9282 CrossRef CAS; (b) W. J. Evans, J. Organomet. Chem., 2002, 647, 2–11 CrossRef CAS.
- (a) P. Jochmann and D. W. Stephan, Chem. Commun., 2014, 50, 8395–8397 RSC; (b) M. Bayram, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2015, 34, 3421–3427 CrossRef CAS.
- C. T. Kiers and A. Vos, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1499–1504 CrossRef.
- K. P. Rao and C. N. R. Rao, Inorg. Chem., 2007, 46, 2511–2518 CrossRef CAS PubMed.
- E. Yang, X.-C. Song, S.-Z. Shen and Y.-D. Lin, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2101 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and data, computational data and crystallographic data. CCDC 1490513–1490514. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc05638e |
| This journal is © The Royal Society of Chemistry 2016 |