
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Consequences of conformational flexibility in hydrogen-bond-driven self-assembly processes†
José Augusto
Berrocal‡
a,
Florent
Di Meo‡
bc,
Miguel
García-Iglesias‡
a,
Ronald P. J.
Gosens
a,
E. W.
Meijer
a,
Mathieu
Linares
*bd and
Anja R. A.
Palmans
*a
aInstitute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
bDepartment of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83 Linköping, Sweden. E-mail: mathieu@ifm.liu.se
cINSERM UMR 850, Univ. Limoges, School of Pharmacy, 2 rue Dr Marcland, F-87025 Limoges, France
dSwedish e-Science Research Centre (SeRC), Linköping University, SE-581 83 Linköping, Sweden
First published on 2nd August 2016
Abstract
We report the synthesis and self-assembly of chiral, conformationally flexible C3-symmetrical trisamides. A strong Cotton effect is observed for the supramolecular polymers in linear alkanes but not in cyclic alkanes. MD simulations suggest 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 conformations of the amides within the aggregates in both types of solvents, but a chiral bias in only linear alkanes.
:1 conformations of the amides within the aggregates in both types of solvents, but a chiral bias in only linear alkanes.
Controlling the nature, directionality and balance of interactions in supramolecular systems is crucial to advance potential applications of supramolecular chemistry.1 Elegant examples have been presented in which molecular design was systematically related to the properties of the superstructures formed.2 In addition, recent progress in the field of living supramolecular polymerizations3 underlined the paramount importance of the conformational entropy of the monomer to yield assemblies of controlled length and narrow dispersity. Besides the monomer design, the solvent also plays a pivotal role in self-assembly processes.4 The chirality, polarity, structure and shape of solvent molecules all affect intra- and intermolecular interactions and thereby the exact nature of the supramolecular assembly.5
The introduction of amides to enable directional hydrogen-bonding interactions in supramolecular motifs has been actively pursued by us and others.6 Due to their easy synthetic accessibility, N,N′,N′′-trialkylbenzene-1,3,5-tricarboxamides (BTAs) were investigated in detail in organic solvents7a and, more recently, in water.7b These molecules form one-dimensional, helical supramolecular polymers, stabilized via threefold intermolecular hydrogen bonding between the amides connected to the aromatic ring. By combining experimental work and theory, the consequences of altering the amide connectivity,8 replacing the amide group by a thioamide9 and separating the amide from the central benzene using rigid, extended, π-conjugated moieties were elucidated.10
Here, we report on the consequences of introducing conformational flexibility into the BTA system by positioning a methylene group between the aromatic ring and the amide moiety, affording methylene-BTAs (mBTAs) (Chart 1). The break of the conjugation allows free rotation between carbon–carbon bonds. We find that such enhanced conformational flexibility significantly increases the cooperative nature of the supramolecular polymerization and affords a self-assembled system in which the expression of supramolecular chirality becomes extremely sensitive to the shape of the alkane solvent. A plausible explanation on the origin of this sensitivity is offered by molecular dynamics simulations.
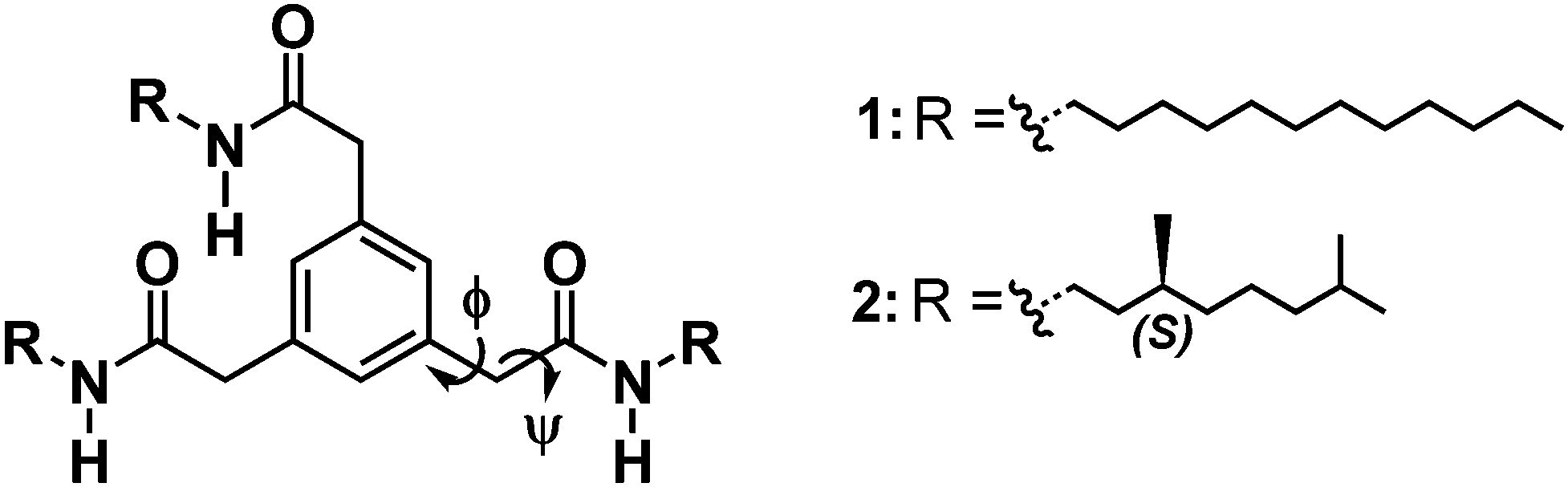 | ||
| Chart 1 Chemical structure of the investigated C3-symmetric methylene-BTAs 1 and 2. | ||
Starting from commercially available 1,3,5-benzenetriacetic acid, mBTAs 1 and 2 were synthesized in two steps in 46% and 72% yield, respectively (ESI†). Achiral mBTA 1 was obtained as a white amorphous solid, whereas chiral 2 was a sticky material. Their bulk characterization was performed using polarized optical microscopy (POM), differential scanning calorimetry (DSC) and temperature-dependent solid state infrared spectroscopy (TD-IR). Cooling both compounds slowly from the isotropic melt afforded pseudo-focal conic textures for 2 in POM, which are typical for columnar liquid crystalline mesophases, whereas no LC behavior was observed for 1 (Fig. S11 and S12, ESI†). The texture of 2 was retained up to 20 °C. DSC measurements showed a sharp transition for both 1 and 2 upon cooling (at 158 °C and 113 °C, respectively), suggesting no morphological transitions in the mesophase of 2 and confirming the higher stability of achiral 1 (Fig. S5 and S10, ESI†).
The presence of hydrogen bonds stabilizing the mesophase was assessed by solid state IR spectroscopy. Diagnostic regions in the IR spectra for intermolecular hydrogen-bond formation are the N–H stretching vibration, the amide carbonyl stretching vibration (amide I) and the combination of C–N stretching and N–H in-plane bending (amide II). For both 1 and 2, ν(N–H) is 3290 cm−1; ν(amide I) is 1645 cm−1 and ν(amide II) is 1550 cm−1 (Fig. 1a and b, Fig. S13 and S14, ESI†). Such vibrations are consistent with the presence of intermolecular hydrogen bonds. Heating the samples above their isotropization temperature resulted in shifting the N–H stretching and the amide I vibration to higher frequencies typical for a loss of hydrogen-bonding interactions (Fig. S13 and S14, ESI†).7a
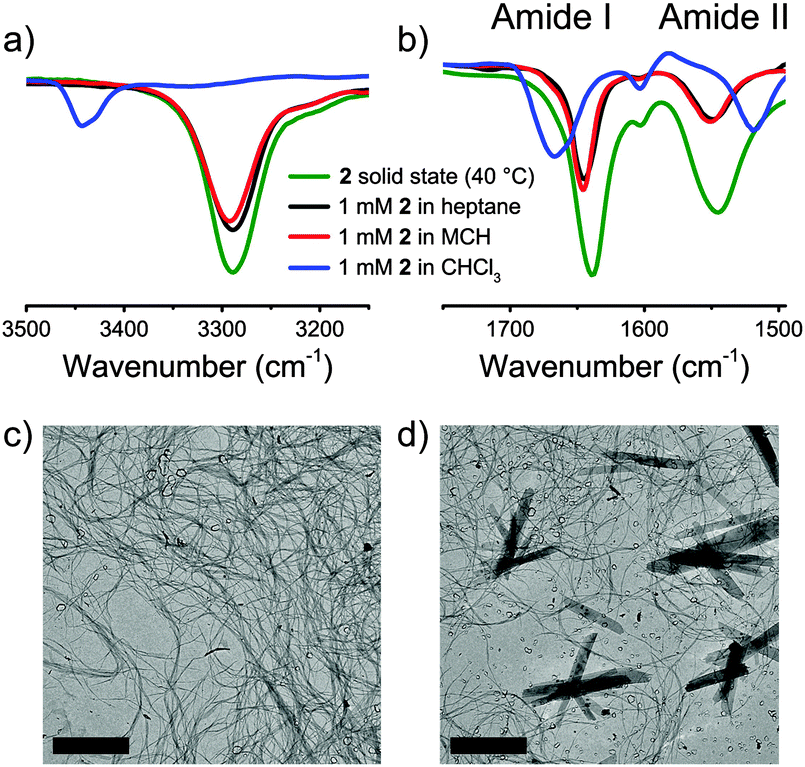 | ||
| Fig. 1 (a) FT-IR (N–H stretching) spectra of 2 in the LC phase (40 °C) and 5 mM solutions; (b) FT-IR (amide I and amide II vibrations) spectra of 2 in the LC phase (40 °C) and 5 mM solutions; TEM micrographs (scale bars 5 μm) from 7.5 mM 2 in (c) heptane and (d) MCH. | ||
Next, we investigated whether the intermolecular hydrogen bonds present in bulk are retained when 1 and 2 are mixed with apolar alkane solvents. The addition of methylcyclohexane (MCH) and heptane to 1 and 2, followed by heating and slow cooling, produced transparent, stiff gels at 5 wt% (66 mM and 74.6 mM for 1 for 2, respectively). To qualitatively assess the organogel-forming ability of mBTAs in alkane solvents, the critical gelation concentration (CGC) was evaluated by vial inversion tests (minimal concentration of mBTA in solution so that no flow occurs upon inverting the vial). Lowering the concentration to 0.5 wt% (6.6 mM for 1 and 7.5 mM for 2) resulted in flowing, yet still viscous, solutions (Fig. S15 and S16, ESI†). Fourier transform infrared spectroscopy (FT-IR) performed on 5 mM solutions of 1 and 2 in MCH and heptane showed spectra similar to those of 2 recorded in bulk at 40 °C, indicating that the hydrogen-bond stabilization of the supramolecular assemblies persists in alkane solvents (Fig. 1a and b, Fig. S17 and S18, ESI†). Differences in the frequencies of the diagnostic regions appear with CHCl3, a solvent in which both 1 and 2 are molecularly dissolved (Fig. 1a and b). The FT-IR spectra also suggested hydrogen-bond-driven self-assembly of 1 and 2 in a number of differently shaped aliphatic solvents (e.g. linear, branched and cyclic alkanes) (Fig. S17 and S18, ESI†). The IR results were further confirmed by temperature-dependent NMR measurements (Fig. S19, ESI†).
While all the evidence collected so far consistently shows the persistence of hydrogen bonding in apolar alkane solvents, remarkable differences appeared when the supramolecular architectures obtained from 7.5 mM (0.5 wt%) solutions of 2 in heptane and MCH were imaged using a transmission electron microscope (TEM) (Fig. 1c and d). In heptane, a dense network of long, thin fibers in the order of tens of micrometers in length and a few nanometers in diameter is observed. Interestingly, in MCH 2 forms shorter fibers of approximately the same diameter, but also ribbon-like architectures in the order of 5–10 μm in length and 0.5–2 μm in width are present (Fig. 1d, Fig. S20 and S21, ESI†).
The combination of IR, NMR, and TEM shows that 1 and 2 form hydrogen-bond stabilized supramolecular assemblies with mostly fibrillar structures. To investigate if mBTAs adopt a helical conformation in which the chiral information embedded in the structure of 2 can be expressed at the supramolecular level, we investigated the aggregates formed by 2 in detail using ultraviolet-visible (UV-Vis) and circular dichroism (CD) spectroscopy in heptane and MCH at 7.5 mM.
The presence of a CD effect is indicative of a bias for one of the helical senses, P or M.11 Although some light scattering was present for the MCH sample, the UV-Vis spectra of 2 in heptane and MCH (7.5 mM) are similar in shape (Fig. 2a), while the CD spectra differ (Fig. 2b). In heptane a strong positive Cotton effect at λmax = 211 nm with a small shoulder at 225 nm is evident. Instead, the CD spectrum of 2 in MCH differs in shape and shows a weak negative Cotton effect at 235 nm.
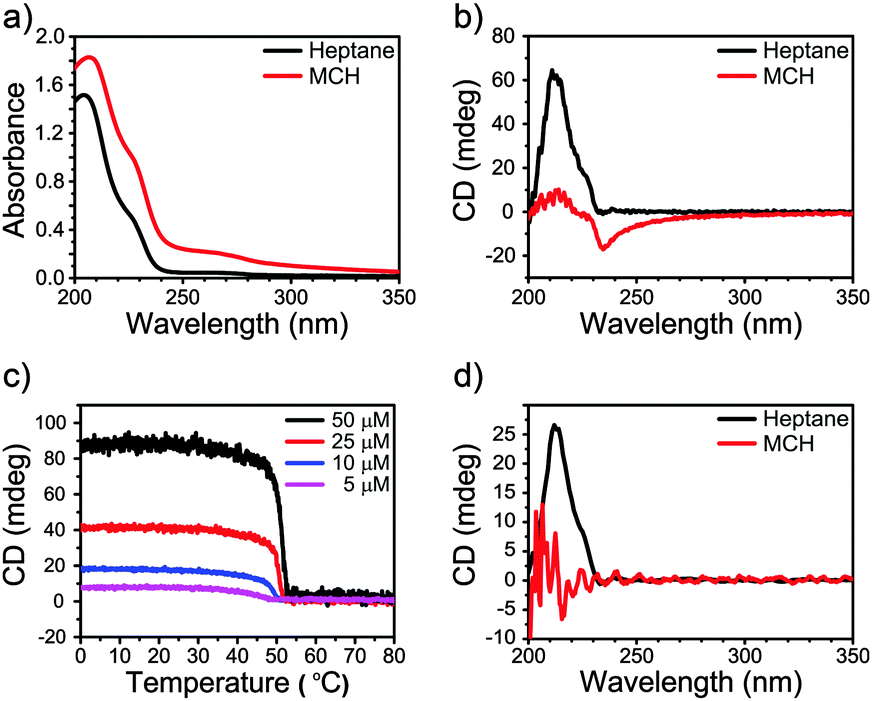 | ||
| Fig. 2 (a) UV-Vis absorption of 2 in heptane and MCH in a high concentration regime (7.5 mM, 20 °C); (b) CD spectra of 2 in heptane and MCH in a high concentration regime (7.5 mM, 20 °C); (c) TD-CD of 2 in heptane (λmax = 211 nm, concentration range 50–5 μM, cooling speed 50 °C h−1); (d) CD spectra of 2 in heptane and MCH in a low concentration regime (20 μM, 20 °C). | ||
In previous investigations, aggregation of supramolecular polymers into higher order aggregates has been shown to affect the shape of the CD spectra.12 To evaluate if higher order aggregation contributes to the observed differences in the CD spectra between heptane and MCH in the case of 2, we performed UV and CD spectroscopy at significantly lower concentrations (50–5 μM). At 100–1000 times lower concentrations, the shapes of the UV spectra (Fig. S22, ESI†) are similar to those of the spectra of 2 at 7.5 mM (Fig. 2a). This suggests that supramolecular assemblies are formed in both solvents also at lower concentrations and higher order aggregation does not play a significant role. In contrast, the CD spectra of 2 at 7.5 mM and 20 μM in heptane were identical in shape, whereas no CD effect was present in MCH. CD spectra were also measured for 2 in other linear/branched and cyclic aliphatic solvents (cyclohexane and decalin, Fig. S23, ESI†) in the same low concentration regime (50–5 μM). In all linear and branched aliphatic solvents a strong, positive Cotton effect was observed, but the use of cyclic solvents always resulted in CD silent spectra.
Finally, temperature-dependent circular dichroism and UV-Vis (TD-CD and TD-UV-Vis, respectively) spectra were measured for solutions of 2 in heptane. The TD-UV-Vis spectra mainly showed an increase in intensity upon increasing the temperature (Fig. S24, ESI†), while the cooling curves obtained for different concentrations showed a very steep increase of the CD effect, indicative of highly cooperative self-assembly (Fig. 2c). The elongation temperature, Te, varies between 55 °C and 45 °C for concentrations between 50 μM and 5 μM. This weak dependence of Te on the concentration indicates a self-assembly process highly dominated by entropy loss.13
The difference in magnitude of the CD effects in cyclic and linear/branched alkanes is remarkable and can have different origins: (i) the slightly higher polarity of the cyclic solvents results in the formation of shorter supramolecular assemblies of 2 in the low concentration regime, (ii) the bias for one helical sense is absent in cyclic solvents under these experimental conditions as a result of differences in the packing of the hydrogen bonds within the supramolecular assemblies, and (iii) the solvent participates in the supramolecular assembly affecting the exact organization of the hydrogen bonds.
Dynamic light scattering (DLS) measurements carried out on 50 μM solutions of 2 in heptane and MCH detected unequivocally larger stacks in the linear solvent (Fig. S26, ESI†). However, the clear resemblance of the UV spectra of 2 in MCH in the different concentration regimes (Fig. 2a and Fig. S22, ESI†) and the identical pattern of the amide vibrations in IR seem to rule out the length of the stacks. No other information about the packing of the hydrogen bonds and solvent–solute interactions could be collected using our available spectroscopic techniques.
To further investigate the self-assembly of 2 molecular dynamics (MD) simulations in both MCH and heptane were performed as well as quantum mechanics (QM) calculations in a vacuum. Two hydrogen bonding networks were considered with either all the amide groups pointing in the same direction (3:0) or with two amides pointing in one direction and the other in the opposite (2:1) (ESI†). P- and M-helices were built with 25 molecules and NPT simulations were run for 40 ns. Conformational analysis was performed on the different MD trajectories to determine the distribution of rise (distance between center of mass of phenyl rings) and twist parameters between adjacent molecules, the distribution of hydrogen bonds, and the distribution of the two dihedral angles Φ and ψ (Chart 1) defining the conformation of the methylene bridge between the phenyl and amide moieties. The different assemblies present very similar distributions of hydrogen bonds and rise despite the two different hydrogen bonding networks (Fig. S27 and S28, ESI†). As expected, we obtain asymmetric distributions of the twist, Φ and ψ for the P- and M-helices, respectively (Fig. S29–S31, ESI†). It is worth noting that for a given helical sense, twist angle distributions show a small but significant population having opposite twist (Fig. S29, ESI†). This underlines the high flexibility of the stacks owing to the methylene bridge as can be seen from the potential energy surface (Fig. S34, ESI†).
MD calculations do not point out a crucial role of solvent in the intrinsic nature of the self-assembly since the conformational parameters exhibit very similar distributions in both MCH and heptane. When looking at the molecule–solvent interactions (Table S1, ESI†) it appears that the 2:1 conformation is more favourable than the 3:0 conformation independently of the solvent. Molecule–molecule and molecule–solvent interactions were weighted and summed to get the total interaction energy per molecule, which also suggests a preference for the 2:1 conformation of the amides upon stacking (Table S1, ESI†). However, very small energy differences are obtained between P- and M-helices featuring the same amide conformation (Table S1, ESI†). Such modest differences can be rationalized with the high flexibility of the monomer and should be considered very carefully as they lie within the margin of accuracy of the method. On the other hand they underline the subtle interplay of interactions that control the self-assembly of conformationally mobile supramolecular monomers.
More convincing evidence for the selection of the 2:1 conformation of the amides upon stacking was obtained from the calculated ECD spectra recorded on snapshots extracted from the MD trajectories (Fig. 3; Fig. S32 and S33, ESI†). The ECD spectrum computed for the 2:1 conformation assembling in a M-helix in heptane (Fig. 3b) presents a very similar shape and sign to those measured experimentally in both high and low concentration regimes (Fig. 2b and d, respectively). Analogously, a stronger CD signal is also calculated for the 2:1 conformation in MCH (Fig. 3d), rather than for the 3:0 conformation (Fig. 3c). The absence of an experimental CD signal at low concentrations in MCH allows us to hypothesize the presence of equimolar amounts of P- and M-helices in the cyclic solvent, as a remarkable demonstration of the sensitivity of the flexible mBTA system to subtle, parametric variations such as the shape of solvent molecules.
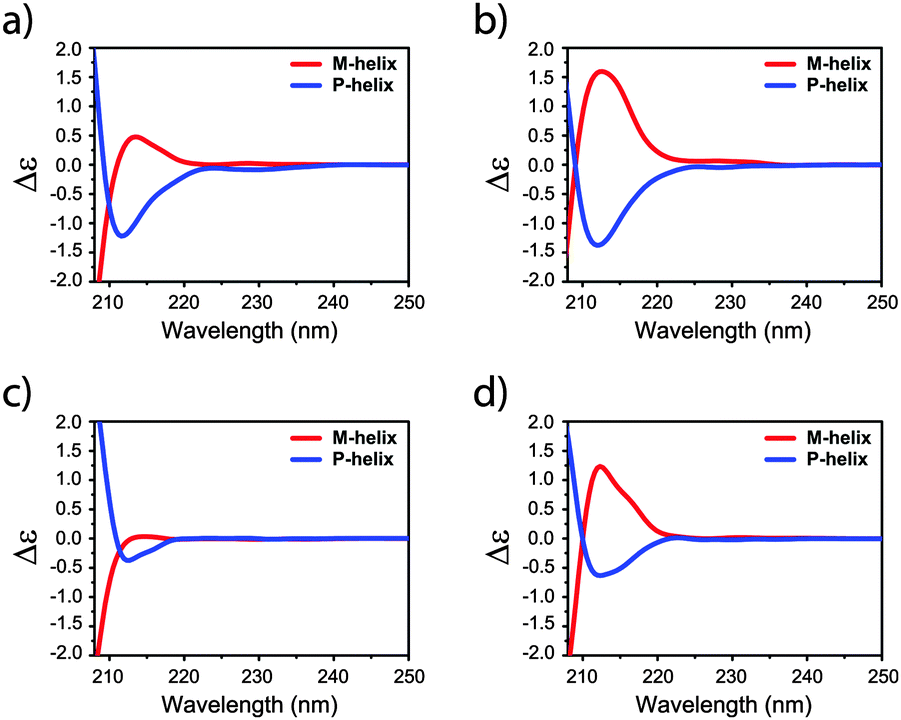 | ||
| Fig. 3 ECD spectra of P- and M-helix of 2 in heptane adopting (a) 3:0 and (b) 2:1 configuration as well as in MCH adopting (c) 3:0 and (d) 2:1 configurations. | ||
In conclusion, in this communication, we have reported the synthesis and self-assembly behavior of mBTAs, similar compounds to the well-studied BTAs. Like BTAs, mBTAs are characterized by a cooperative hydrogen-bond-driven self-assembly. Tiny macroscopic differences are observed between the supramolecular polymers in heptane and MCH. In stark contrast, spectroscopic studies reveal the preference for a helical sense in linear alkane solvents and the absence of a chiral bias in cyclic ones. In line with previous investigations,4c we conclude that the solvent plays a dominant role when the difference in the intrinsic energy between P and M-helices is small. Our communication emphasizes the pivotal role of molecular design and solvent–solute interactions in the self-assembly behavior of supramolecular monomers. More specifically, it highlights the necessity to comprehend the role of each molecular component in self-assembly. Only by doing that, perfect control and predictability in one-dimensional supramolecular polymerizations can be achieved.
JAB, MGI, RPAG, EWM and ARAP would like to thank the Gravity program 024.001.035, NWO TOP-PUNT 718.014.003 for financial support and Anneloes Oude Vrielink for TEM imaging. FDM and ML acknowledge the Swedish e-Research Center (SeRC) for financial support, the Swedish Research Council (Grant No. 621-2014-4646), SNIC (Swedish National Infrastructure for Computing) and Dr Julien Idé for providing the code for exciton coupling calculations.
Notes and references
- T. Aida, E. W. Meijer and S. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- (a) H. Sun, C. A. Hunter and E. M. Llamas, Chem. Sci., 2015, 6, 1444 RSC; (b) S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507 CrossRef CAS; (c) E. Busseron, Y. Ruff, E. Moulina and N. Giuseppone, Nanoscale, 2013, 5, 7098 RSC; (d) A. Jain and S. J. George, Mater. Today, 2015, 18, 206 CrossRef CAS; (e) W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634 CrossRef CAS PubMed; (f) F. Pop, C. Melan, I. Danila, M. Linares, D. Belijonne, D. B. Amabilino and N. Avarvari, Chem. – Eur. J., 2014, 20, 17443 CrossRef CAS PubMed.
- (a) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed; (b) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS PubMed; (c) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed; (d) R. Dev Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241 CrossRef PubMed.
- (a) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80 CrossRef CAS PubMed; (b) J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. C. Vinter, Angew. Chem., Int. Ed., 2007, 46, 3706 CrossRef CAS PubMed; (c) S. Cantekin, Y. Nakano, J. C. Everts, P. van der Schoot, E. W. Meijer and A. R. A. Palmans, Chem. Commun., 2012, 48, 3803 RSC; (d) K. S. Mali, K. Lava, K. Binnemans and S. De Feyter, Chem. – Eur. J., 2010, 16, 14447 CrossRef CAS PubMed.
- (a) D. Dasgupta, A. Thierry, C. Rochas, A. Ajayaghosh and J. M. Guenet, Soft Matter, 2012, 8, 8714 RSC; (b) N. Ponnuswamy, G. Dan Pantoş, M. M. J. Smulders and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 566 CrossRef CAS PubMed; (c) M. Raynal and L. Boutellier, Chem. Commun., 2011, 47, 8271 RSC; (d) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421 CrossRef CAS PubMed; (e) C. A. Khatri, Y. Pavlova, M. M. Green and H. Morawetz, J. Am. Chem. Soc., 1997, 119, 6991 CrossRef CAS; (f) A. L. Hofacker and J. R. Paquette, Angew. Chem., Int. Ed., 2005, 44, 1053 CrossRef CAS PubMed; (g) B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni and L. Boutellier, Chem. – Eur. J., 2010, 16, 173 CrossRef CAS PubMed; (h) V. V. Borovkov, G. A. Hembury and Y. Inoue, Acc. Chem. Res., 2004, 37, 449 CrossRef CAS PubMed.
- (a) J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278 CrossRef CAS PubMed; (b) D. Miyajima, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, Angew. Chem., Int. Ed., 2011, 50, 7865 CrossRef CAS PubMed; (c) J. Guilleme, M. J. Mayoral, J. Calbo, J. Aragó, P. M. Viruela, E. Ortí, T. Tomas and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 2543 CrossRef CAS PubMed; (d) X.-Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871 RSC; (e) F. Helmich, C. C. Lee, M. M. L. Nieuwenhuizen, J. C. Gielen, P. C. M. Christianen, A. Larsen, G. F. Fytas, P. E. L. G. Leclère, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2010, 49, 3939 CrossRef CAS PubMed; (f) M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekemans, A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336 CrossRef CAS PubMed; (g) I. Destoop, H. Xu, C. Oliveras-González, E. Ghijsens, D. B. Amabilino and S. De Feyter, Chem. Commun., 2013, 49, 7477 RSC.
- (a) S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC; (b) C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963 RSC.
- P. J. M. Stals, J. Everts, R. de Bruijn, I. A. W. Filot, M. M. J. Smulders, R. Martín-Rapún, E. A. Pidko, T. F. A. de Greef, A. R. A. Palmans and E. W. Meijer, Chem. – Eur. J., 2010, 16, 810 CrossRef CAS PubMed.
- T. Mes, S. Cantekin, D. W. R. Balkenende, M. M. M. Frissen, M. A. J. Gillissen, B. F. M. De Waal, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2013, 19, 8642 CrossRef CAS PubMed.
- F. García, P. A. Korevaar, A. Verlee, E. W. Meijer, A. R. A. Palmans and L. Sánchez, Chem. Commun., 2013, 49, 8674 RSC.
- D. A. Ligthner and J. E. Gurst, Organic Conformational Analysis And Stereochemistry From Circular Dichroism Spectroscopy, Wiley-VCH, New York, 2000 Search PubMed.
- (a) P. J. M. Stals, P. A. Korevaar, M. A. J. Gillissen, T. F. A. de Greef, C. F. C. Fitié, R. P. Sijbesma, A. R. A. Palmans and E. W Meijer, Angew. Chem., Int. Ed., 2012, 51, 11297 CrossRef CAS PubMed; (b) X. Li, L. Zhu, S. Duan, Y. Zhao and H. Ågren, Phys. Chem. Chem. Phys., 2014, 16, 23854 RSC; (c) Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed., 2014, 53, 13424 CrossRef CAS PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis and characterization of 1 and 2, POM images of 1 and 2, solid state TD-IR measurements, gelation studies, solution phase IR, self-assembly of 2 in deuterated MCH, TEM micrographs of supramolecular polymers in heptane and MCH, UV-Vis and CD spectra of 2 in different alkane solvents, DLS experiments and computational details. See DOI: 10.1039/c6cc05593a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |