
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Replication of α-amino acids via Strecker synthesis with amplification and multiplication of chiral intermediate aminonitriles†
Shohei
Aiba
,
Naoya
Takamatsu
,
Taichiro
Sasai
,
Yuji
Tokunaga
and
Tsuneomi
Kawasaki
*
Department of Materials Science and Engineering, University of Fukui, Bunkyo, Fukui 910-8507, Japan. E-mail: tk@u-fukui.ac.jp
First published on 28th July 2016
Abstract
Replication of chiral L- and D-α-(p-tolyl)glycine has been achieved in combination with the asymmetric induction, amplification and multiplication of their own chiral intermediates, L- and D-aminonitriles, in the solid-phase via the Strecker reaction between three achiral components, which is a plausible prebiotic mechanism for amino acid synthesis.
The origin and amplification of chirality leading to biological homochirality, as exemplified by the L-amino acids and D-sugars, have attracted much attention. Starting from the discovery of molecular chirality,1 original research studies2 on the origin of chirality have been conducted including the effect of chiral physical factors3 such as circularly polarized light,4 the chiral surface of inorganic crystals5 such as quartz, chiral crystallization of achiral compounds6 including stereospecific solid-state reactions2d,7 and the spontaneous absolute asymmetric synthesis.8 The induced small chirality by the suggested mechanisms should be significantly enhanced toward high enantioenrichment as seen in biological compounds by the appropriate mechanisms9 including the phenomenon of self-disproportionation of enantiomers.10 Asymmetric autocatalysis with amplification of enantiomeric excess (ee)2e,11 should be strongly correlated with the homochirality of organic compounds. In addition, it has been reported that chiral amino acids including meteoritic compounds12 could act as a source of chirality to synthesize the enantioenriched compounds13 such as sugars and related compounds.14
On the other hand, the Strecker reaction15 (Fig. 1) has long been considered as one of the methods for the synthesis of α-amino acids on primitive Earth before the origin of life.16 Therefore, replicative generation of highly enantioenriched amino acids via this mechanism is an emerging research subject in the fields of prebiotic, synthetic, and systems chemistry17 and could be one of the possible approaches to achieving biological homochirality.
 | ||
| Fig. 1 Concept of the present research: replicative Strecker amino acid synthesis. | ||
Here we report the selective formation of a nearly enantiomerically pure α-aminonitrile, which is a chiral intermediate of the Strecker amino acid synthesis. It is produced by the reaction between hydrogen cyanide (HCN), a carbonyl compound and an amine. We found that an amino acid could induce the enantiomeric imbalance toward the formation of its own chiral intermediate (Fig. 1). Therefore, as a result of the combination of the amplification and multiplication of aminonitrile, a more near enantiopure amino acid could be newly synthesized after the hydrolysis of aminonitrile. The present reactions demonstrate one of the models in which the chiral intermediate α-aminonitrile plays a key role in the generation and evolution of homochirality of amino acids via the Strecker synthesis.
We have previously reported18 the formation of enantioenriched α-(p-tolyl)glycine nitrile 1via spontaneous crystallization8b,19 after a three-component Strecker reaction between achiral HCN, p-tolualdehyde (3) and benzhydrylamine (4). The stochastic distribution in the formation of L- and D-enriched enantiomorphs 1 was observed. In this work, the formation of a selected enantiomer between L- and D-α-(p-tolyl)glycine nitrile 1 was achieved by using chiral amino acids, i.e., L- and D-α-(p-tolyl)glycine (2), as a source of chirality (Fig. 2). As the enantiomeric imbalance of 1 induced by amino acid 2 can be significantly enhanced by the dissolution/crystallization-mediated amplification of ee,20,21 and the amount of enantioenriched 1 can also be multiplied, a large amount of near enantiomerically pure amino acid 2, with the same molecular handedness as that of the original, could be newly synthesized in a highly enantioselective manner after the hydrolysis of aminonitrile 1.
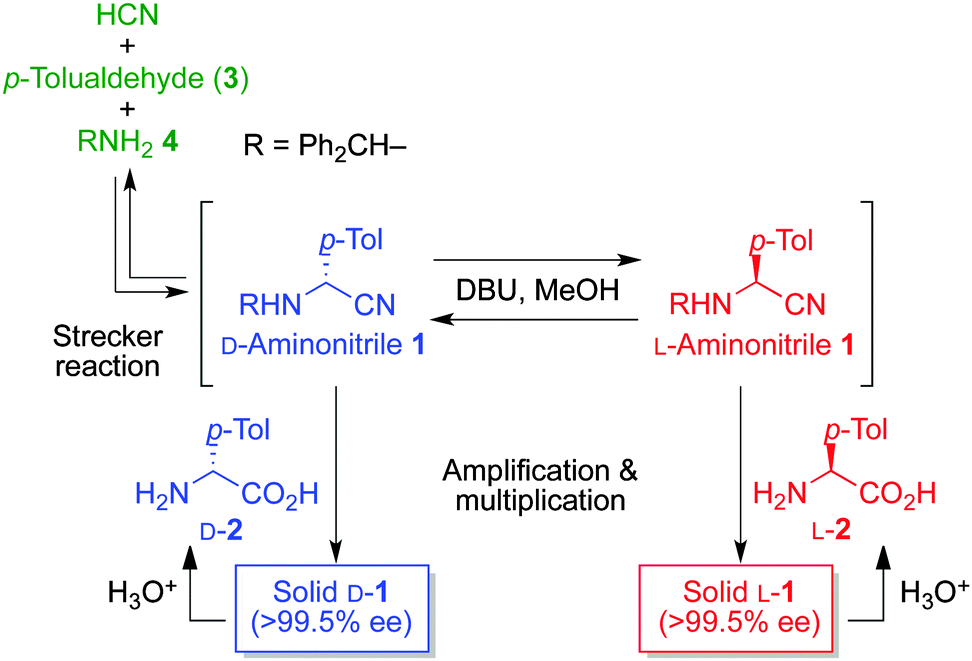 | ||
| Fig. 2 Replicative formation of L- and D-α-(p-tolyl)glycine (2) via asymmetric induction in the formation of an enantioenriched conglomerate of aminonitrile 1. | ||
In the presence of L- or D-α-(p-tolyl)glycine (2), the formation and amplification of enantiomorphic aminonitrile 1 were performed via a Strecker reaction between three achiral substrates: HCN, aldehyde 3, and amine 4 (Fig. 2 and Table 1). When highly enantioenriched L-2 with 97% ee was used as a source of chirality, L-aminonitrile 1 with >99.5% ee was isolated in 45% yield by filtration after the amplification of the solid-phase ee (entry 1). Near racemic 1 was isolated from the solution-phase (filtrate) in 24% yield. On the other hand, D-amino acid 2 led to the formation of near enantiopure D-1 after four amplification cycles (entry 2). No other compounds such as the product of benzoin condensation of p-tolualdehyde could be isolated from the filtrate except for highly polar materials.
| Entrya | Amino acid 2 | Aminonitrile 1 | |||
|---|---|---|---|---|---|
| % ee | Config. | % eeb | Config. | % Yieldc | |
a The molar ratio used was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4:HCN = 1:2:2:3 (mmol). The reactions were performed in a 1.0 M DBU solution in methanol in the presence of a stir bar without using glass beads. Amino acid 2 completely dissolves in the reaction medium containing DBU.
b The ee value was determined by high performance liquid chromatography (HPLC), employing a chiral stationary phase.
c The isolated yield of solid product 1 by filtration.
d
L-1 with 75% ee was obtained after four cycles of amplification; two additional cycles enhanced the ee to >99.5% ee. Near racemic 1 was isolated from the solution-phase (filtrate) in 24% yield.
e
D-1 with 71% ee was obtained after three cycles of amplification; one additional cycle enhanced the ee to >99.5%. Near racemic 1 was isolated from the solution-phase (filtrate) in 28% yield.
f Eight cycles of amplification of solid-phase ee were performed.
g Eleven cycles of amplification of solid-phase ee were performed.
h The thermal cycle was started after the dissolution of amino acid 2 in a suspension of racemic conglomerate 1. :3:4:HCN = 1:2:2:3 (mmol). The reactions were performed in a 1.0 M DBU solution in methanol in the presence of a stir bar without using glass beads. Amino acid 2 completely dissolves in the reaction medium containing DBU.
b The ee value was determined by high performance liquid chromatography (HPLC), employing a chiral stationary phase.
c The isolated yield of solid product 1 by filtration.
d
L-1 with 75% ee was obtained after four cycles of amplification; two additional cycles enhanced the ee to >99.5% ee. Near racemic 1 was isolated from the solution-phase (filtrate) in 24% yield.
e
D-1 with 71% ee was obtained after three cycles of amplification; one additional cycle enhanced the ee to >99.5%. Near racemic 1 was isolated from the solution-phase (filtrate) in 28% yield.
f Eight cycles of amplification of solid-phase ee were performed.
g Eleven cycles of amplification of solid-phase ee were performed.
h The thermal cycle was started after the dissolution of amino acid 2 in a suspension of racemic conglomerate 1.
|
|||||
| 1d | 97 | L | >99.5 | L | 45 |
| 2e | 97 | D | >99.5 | D | 44 |
| 3 | ca. 50 | L | 99 | L | 42 |
| 4f | ca. 50 | D | 98 | D | 48 |
| 5g | ca. 10 | L | >99.5 | L | 27 |
| 6 | ca. 10 | D | >99.5 | D | 38 |
| 7h | ca. 50 | L | 75 | L | 21 |
| 8h | ca. 50 | D | 47 | D | 20 |
The enantiomeric purity of suspended solid 1 was enhanced by the subsequent heating/cooling cycles. After partial dissolution of suspended solid 1 (ca. 80–90%) in the reaction mixture at 45–50 °C, the remaining conglomerate 1 regrew during the gradual cooling to room temperature over a period of 1 hour. The repetition of this thermal dissolution/crystallization cycle amplified the ee of solid 1 significantly to afford 1 with up to >99.5% ee.
Even when L- and D-2 with as low as approximately 50 and 10% ee were provided as chiral triggers, highly enantioenriched L- and D-solids 1 could be isolated with the same stereochemical relationships, respectively (entries 3–6). Therefore, L-amino acid 2 induced the production of L-aminonitrile 1 and D-2 initiated the propagation of D-1. When L- and D-2 were dissolved in a prepared suspension of racemic conglomerate 1, crystalline solid 1 acquired L- and D-excess, respectively, after an adequate number of thermal cycles (entries 7 and 8). Stereoselective adsorption22 of amino acid 2 at the crystal surface of conglomerate 1 would be included as a source of asymmetric induction and amplification, which subsequently differentiates the rate of crystal growth and dissolution between L- and D-enantiomorphs 1. As enantioenriched 1 can be hydrolyzed to 2 without a decrease in enantiopurity,18 and the molecular handedness of the source and product is the same, replication of amino acid 2 is subsequently achieved.
It should be noted that amino acids other than α-(p-tolyl)glycine (2) could also work as chiral triggers for the formation of enantioenriched 1 (Table 2). Therefore, L-aminonitrile 1 was formed by the Strecker reaction between HCN, aldehyde 3 and primary amine 4 in the presence of L-alanine after the amplification of solid-phase ee (entry 1). Thus, the opposite D-alanine induced the formation of D-1 (entry 2). L- and D-Phenylalanine also acted as a source of chirality for the production of L- and D-1, respectively (entries 3 and 4). When valine was subjected to this reaction, L-enantiomer affords D-1 and D-valine gave the L-enantiomorph of 1 (entries 5 and 6). Unnatural phenylglycine that is a demethyl derivative of 2 also worked as an efficient chiral trigger to produce 1 (entries 7 and 8). In these reactions, chiral inducers, i.e., amino acids completely dissolved in the reaction medium including DBU same as in the case when using L- and D-2.
|
|
||||
|---|---|---|---|---|
| Entrya | Amino acid | Aminonitrile 1b | ||
| % ee | Config. | % Yield | ||
|
a The molar ratio used was amino acid:3:4:HCN = 1:2:2:3 (mmol). The reactions were performed in a 1.0 M DBU solution in methanol. All amino acids completely dissolve in the reaction medium containing DBU.
b The ee value was determined by HPLC, employing a chiral stationary phase.
|
||||
| 1 | L-Alanine | 92 | L | 54 |
| 2 | D-Alanine | >99.5 | D | 50 |
| 3 | L-Phenylalanine | 99 | L | 54 |
| 4 | D-Phenylalanine | 99 | D | 49 |
| 5 | L-Valine | 99 | D | 58 |
| 6 | D-Valine | 99 | L | 45 |
| 7 | L-Phenylglycine | 99 | L | 41 |
| 8 | D-Phenylglycine | 98 | D | 32 |
Next, we demonstrate the amplification of solid-phase ee from extremely low ee (ca. 0.05% ee) to a near enantiopure state (Fig. 3 and Table S1, ESI†). Solid 1 with the indicated ee was prepared by mixing racemic conglomerate 1 and near enantiopure L- or D-conglomerates 1, and the resulting 1 (with indicated ee) was suspended in a 1 M DBU solution in methanol in the presence of a stir bar without glass beads. This mixture was then subjected to repeated dissolution/crystallization-induced amplification as mentioned above. When L-1 with ca. 5% ee was subjected to the thermal cycle, the ee was amplified to 26% ee (L). The next cycle afforded L-solid 1 with 91% ee; finally, >99.5% ee (L) was achieved. The recovered yield of 1 was 54%. The opposite D-1 with ca. 5% ee could also be amplified to 96% ee (D) after four thermally controlled amplifications. Even when the enantioenrichment was reduced to ca. 0.5% ee, the initially existing major enantiomorph 1 dominated; up to >99.5% ee was achieved after five heating/cooling cycles. Furthermore, the initial very small L- and D-imbalances, approximately 0.05% ee, were also significantly enhanced during five amplification cycles; up to >99.5% ee was achieved.
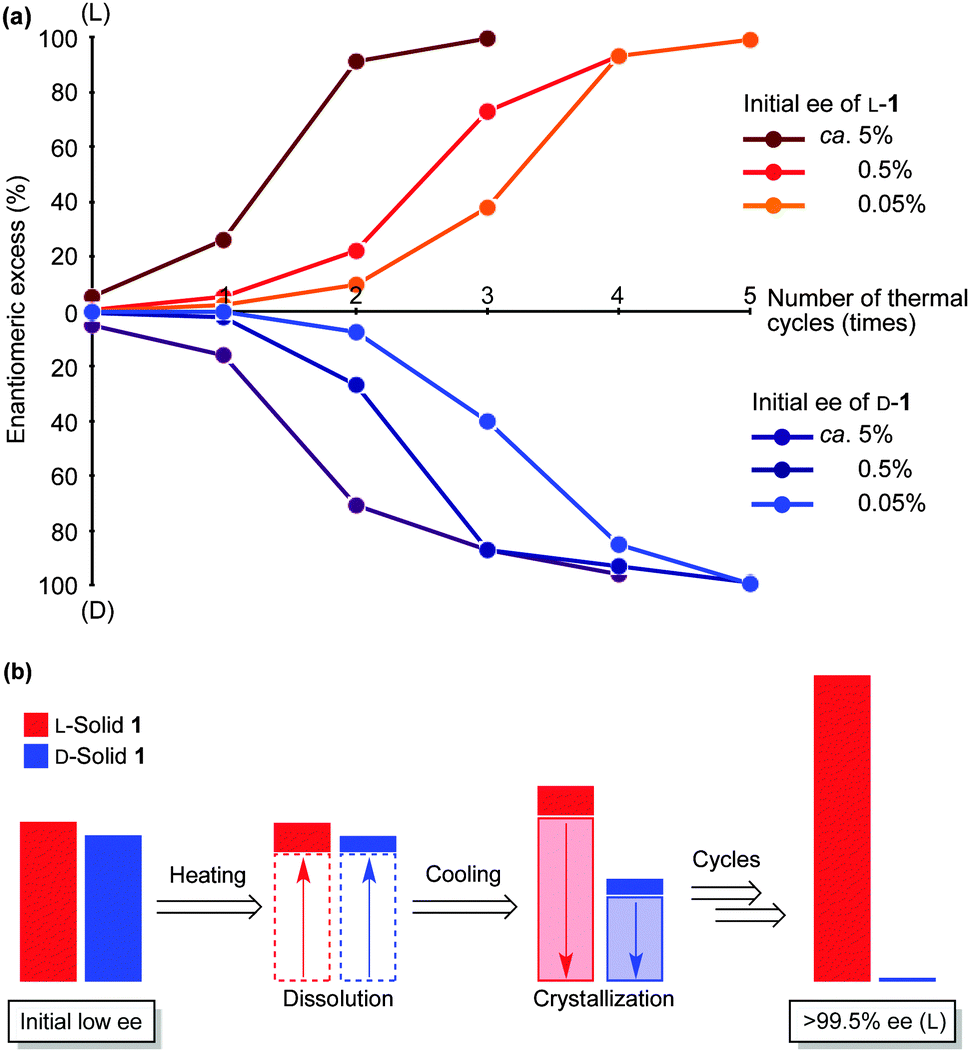 | ||
| Fig. 3 (a) Amplification of solid-phase ee of L- and D-α-aminonitrile 1 from extremely low ee (ca. 0.05% ee) to near enantiopurity (>99.5% ee) by the heating/cooling cycles. (b) Simplified model for the production of highly enantioenriched solid 1 (L-enantiomorph). | ||
A linear relationship was observed between the initial ee and the number of thermal cycles required to achieve high ee, therefore, it was assumed that near racemate 1 dissolved during the heating step to afford a reduced amount of suspended solid 1 with amplified ee (Fig. 3b). In turn, cooling-induced deracemization may occur during gradual crystal growth without a decrease in the amplified ee. In the simple calculations (Fig. S1, ESI†), dissolution of 80% of rac-1 from enantioenriched solid 1 with 5% ee (enantiomeric ratio (er) = 52.5/47.5) affords solid 1 with 25% ee (er = 12.5/7.5). After the ideal deracemization of 1 (er = 62.5/37.5, 25% ee), the next cycle affords a near enantiomerically pure solid (er = 100/0, 100% ee). It was calculated that four and five thermal cycles afford enantiopure solids starting from solid 1 with 0.5% ee and 0.05% ee, respectively. Since the initial very low ee (ca. 0.05%) could be enhanced to become a near enantiopure state, it was supposed that exact racemic dissolution during the heating step and efficient deracemization during the cooling step are key to this process. Therefore, this practically perfect thermal cycle is a highly sensitive and powerful method to improve an extremely small imbalance of L- and D-enantiomorphs of 1 toward a near enantiopure state (>99.5% ee).
In addition, reactive crystallization of selected enantiomer 1 was realized; thus, seed crystal 1 gave a further amount of a near enantiomerically pure crystalline solid 1 by the portion-wise addition of three achiral reagents (Fig. 4 and Table S2, ESI†). For example, using L-1 (0.08 g) with >99.5% ee as a seed, five consecutive additions of HCN, aldehyde 3, and amine 4 were conducted to precipitate L-1 (46.5 g) with >99.5% ee in 76% isolated yield, without thermal amplification. On the other hand, the amount of D-1 with >99.5% ee could also be increased as a result of the three-component Strecker reaction. Therefore, automultiplication of near enantiopure L- and D-α-aminonitrile 1 has been achieved in a highly stereospecific manner. To the best of our knowledge, this is the first example of highly enantioselective reactive crystallization via a three component reaction involving C–C bond formation.
 | ||
| Fig. 4 Amplification of solid-phase ee of L- and D-α-aminonitrile 1 from extremely low ee (ca. 0.05% ee) to near enantiopurity (>99.5% ee) by the heating/cooling cycles. | ||
In summary, we have demonstrated one of the reaction models for the replication of chiral α-amino acids via the proposed prebiotic mechanism—the Strecker synthesis. Amino acids, i.e., L- and D-α-(p-tolyl)glycine (2), acting as a source of chirality, asymmetric induction, amplification, and multiplication, were realized at the formation stage of the chiral intermediate α-aminonitrile 1, and amino acid 2 with the same structure and configuration as those of the original amino acid was newly synthesized by the subsequent hydrolysis. Even if a small amount of amino acid with low ee was induced by the proposed origin of chirality initially, it is possible to obtain both quantitatively and enantiomerically enhanced amino acids by the reaction sequence discussed here. These results should contribute to one of the approaches toward understanding the origin and amplification of homochirality such as seen in L-amino acids.
This work was supported by JSPS KAKENHI, the Takeda Science Foundation and Cooperative Program of Advanced Medicine and Engineering Research of University of Fukui. T. K. thanks the Research Center for Chirality, Research Institute for Science & Technology, Tokyo University of Science.
Notes and references
- L. Pasteur, Ann. Chim. Phys., 1848, 24, 442 Search PubMed.
- (a) J. L. Bada, Nature, 1995, 374, 594 CrossRef CAS PubMed; (b) M. Bolli, R. Micura and A. Eschenmoser, Chem. Biol., 1997, 4, 309 CrossRef CAS PubMed; (c) J. S. Siegel, Chirality, 1998, 10, 24 CrossRef CAS; (d) I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236 CrossRef CAS PubMed; (e) K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643 CrossRef CAS PubMed.
- (a) J. M. Ribó, J. Crusats, F. Sagues, J. Claret and R. Rubires, Science, 2001, 292, 2063 CrossRef PubMed; (b) T. Kawasaki, Y. Matsumura, T. Tsutsumi, K. Suzuki, M. Ito and K. Soai, Science, 2009, 324, 492 CrossRef CAS PubMed.
- (a) A. Moradpour, J. F. Nicoud, G. Balavoine, H. B. Kagan and G. Tsoucaris, J. Am. Chem. Soc., 1971, 93, 2353 CrossRef; (b) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418 CrossRef; (c) Y. Inoue, Chem. Rev., 1992, 92, 741 CrossRef CAS; (d) T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274 CrossRef CAS PubMed; (e) W. L. Noorduin, A. A. C. Bode, M. van der Meijden, H. Meekes, A. F. van Etteger, W. J. P. van Enckevort, P. C. M. Christianen, B. Kaptein, R. M. Kellogg, T. Rasing and E. Vlieg, Nat. Chem., 2009, 1, 729 CrossRef CAS PubMed.
- (a) R. M. Hazen and D. S. Sholl, Nat. Mater., 2003, 2, 367 CrossRef CAS PubMed; (b) W. A. Bonner, P. R. Kavasmaneck, F. S. Martin and J. J. Flores, Science, 1974, 186, 143 CAS; (c) K. Soai, S. Osanai, K. Kadowaki, S. Yonekubo, T. Shibata and I. Sato, J. Am. Chem. Soc., 1999, 121, 11235 CrossRef CAS.
- D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975 CAS.
- (a) M. Sakamoto, Chem. – Eur. J., 1997, 3, 684 CrossRef CAS; (b) T. Matsuura and H. Koshima, J. Photochem. Photobiol., C, 2005, 6, 7 CrossRef CAS.
- (a) K. Mislow, Collect. Czech. Chem. Commun., 2003, 68, 849 CrossRef CAS; (b) E. Havinga, Biochim. Biophys. Acta, 1954, 13, 171 CrossRef CAS PubMed; (c) K. Soai, I. Sato, T. Shibata, S. Komiya, M. Hayashi, Y. Matsueda, H. Imamura, T. Hayase, H. Morioka, H. Tabira, J. Yamamoto and Y. Kowata, Tetrahedron: Asymmetry, 2003, 14, 185 CrossRef CAS; (d) D. A. Singleton and L. K. Vo, Org. Lett., 2003, 5, 4337 CrossRef CAS PubMed.
- (a) T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef CAS PubMed; (b) M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138 CrossRef; (c) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed; (d) M. Klussmann, H. Iwamura, S. P. Mathew, D. H. Wells Jr, U. Pandya, A. Armstrong and D. G. Blackmond, Nature, 2006, 441, 621 CrossRef CAS PubMed; (e) Y. Hayashi, M. Matsuzawa, J. Yamaguchi, S. Yonehara, Y. Matsumoto, M. Shoji, D. Hashizume and H. Koshino, Angew. Chem., Int. Ed., 2006, 45, 4593 CrossRef CAS PubMed; (f) R. Breslow and M. S. Levine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12979 CrossRef CAS PubMed; (g) W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600 CrossRef CAS PubMed.
- (a) V. A. Soloshonok, C. Roussel, O. Kitagawa and A. E. Sorochinsky, Chem. Soc. Rev., 2012, 41, 4180 RSC; (b) V. A. Soloshonok, H. Ueki, M. Yasumoto, S. Mekala, J. S. Hirschi and D. A. Singleton, J. Am. Chem. Soc., 2007, 129, 12112 CrossRef CAS PubMed; (c) T. Katagiri, C. Yoda, K. Furuhashi, K. Ueki and T. Kubota, Chem. Lett., 1996, 115 CrossRef CAS.
- (a) F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459 CrossRef CAS PubMed; (b) K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS.
- J. R. Cronin and S. Pizzarello, Science, 1997, 275, 951 CrossRef CAS PubMed.
- T. Shibata, J. Yamamoto, N. Matsumoto, S. Yonekubo, S. Osanai and K. Soai, J. Am. Chem. Soc., 1998, 120, 12157 CrossRef CAS.
- (a) S. Pizzarello and A. L. Weber, Science, 2004, 303, 1151 CrossRef CAS PubMed; (b) A. Córdova, M. Engqvist, I. Ibrahem, J. Casas and H. Sundén, Chem. Commun., 2005, 2047 RSC; (c) R. Breslow and Z.-L. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5723 CrossRef CAS PubMed; (d) J. E. Hein, E. Tse and D. G. Blackmond, Nat. Chem., 2011, 3, 704 CrossRef CAS PubMed.
- A. Strecker, Ann. Chem. Pharm., 1850, 75, 27 CrossRef.
- (a) S. L. Miller, Science, 1953, 117, 528 CAS; (b) J. L. Bada, Chem. Soc. Rev., 2013, 42, 2186 RSC.
- (a) K. R. Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285 CrossRef PubMed; (b) D. Sievers and G. von Kiedrowski, Nature, 1994, 369, 221 CrossRef CAS PubMed; (c) D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525 CrossRef CAS PubMed.
- T. Kawasaki, N. Takamatsu, S. Aiba and Y. Tokunaga, Chem. Commun., 2015, 51, 14377 RSC.
- (a) F. Yagishita, H. Ishikawa, T. Onuki, S. Hachiya, T. Mino and M. Sakamoto, Angew. Chem., Int. Ed., 2012, 51, 13023 CrossRef CAS PubMed; (b) R. R. E. Steendam, J. M. M. Verkade, T. J. B. van Benthem, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Nat. Commun., 2014, 5, 5543 CrossRef CAS PubMed.
- (a) C. Viedma, J. E. Ortiz, T. de Torres, T. Izumi and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 15274 CrossRef CAS PubMed; (b) W. L. Noorduin, T. Izumi, A. Mille Maggi, M. Leeman, H. Meekes, W. J. P. van Enckevort, R. M. Kellogg, B. Kaptein, E. Vlieg and D. G. Blackmond, J. Am. Chem. Soc., 2008, 130, 1158 CrossRef CAS PubMed; (c) C. Hongo, M. Tohyama, R. Yoshioka, S. Yamada and I. Chibata, Bull. Chem. Soc. Jpn., 1985, 58, 433 CrossRef CAS.
- (a) K. Suwannasang, A. E. Flood, C. Rougeot and G. Coquerel, Cryst. Growth Des., 2013, 13, 3498 CrossRef CAS; (b) R. R. E. Steendam, T. J. B. van Benthem, E. M. E. Huijs, H. Meekes, W. J. P. van Enckevort, J. Raap, F. P. J. T. Rutjes and E. Vlieg, Cryst. Growth Des., 2015, 15, 3917 CrossRef CAS.
- (a) L. Addadi, Z. Berkovitch-Yellin, N. Domb, E. Gati, M. Lahav and L. Leiserowitz, Nature, 1982, 296, 21 CrossRef CAS; (b) L. Addadi, S. Weinstein, E. Gati, I. Weissbuch and M. Lahav, J. Am. Chem. Soc., 1982, 104, 4610 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and details of experimental results. See DOI: 10.1039/c6cc05544c |
| This journal is © The Royal Society of Chemistry 2016 |