
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability and reactivity control of carbenoids: recent advances and perspectives
Viktoria H.
Gessner

Inorganic Chemistry II – Organometallic Chemistry, Faculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstraße 150, D-44801 Bochum, Germany. E-mail: vgessner@rub.de; Fax: +49 (0)234 32 14174; Tel: +49 (0)234 32 24174
First published on 1st August 2016
Abstract
Metal carbenoids such as lithium or Simmons–Smith-type reagents are widely used in organic synthesis, particularly in cyclopropanation and homologation reactions. These reagents are often highly reactive and thermally labile, thus limiting their isolation and hampering the development of new synthetic applications. Recent years however, have shown that by means of systematic stabilization a control of reactivity and the development of new applications is possible. This feature article documents recent developments in the control of carbenoid reactivity and stability and highlights structural and electronic properties as well as applications in main group element and transition metal chemistry.
 Viktoria H. Gessner | Viktoria Gessner (born Oct. 1982 in Würzburg) obtained her diploma degree in 2007 from the University of Würzburg and her PhD in 2009 from the TU Dortmund working with C. Strohmann. After a postdoctoral stay at UC Berkeley with T. D. Tilley she started her independent academic career at the University of Würzburg affiliated to the group of H. Braunschweig and finished her Habilitation in 2015. She recently moved to the Ruhr-Universität Bochum, where she is now chair for inorganic chemistry. Her research interests involve organometallic and main group metal chemistry with focus on s-block metal compounds and unique carbon-based ligand systems. Her work has been recognized by several grants and awards, such as by an Emmy Noether Grant of the German Research Council, a Starting Grant of the European Research Council or the ADUC award and IUPAC prize for young scientists. |
Introduction
In recent years, research in main group element and metal chemistry has experienced a remarkable renaissance due to their potential use in bond activation chemistry and catalysis.1 Particularly low-valent main group species with vacant coordination sites and small energy gaps between their frontier orbitals have shown to be capable of transformations, which have long been considered the realm of transition metals.2 The advancements in carbene chemistry have especially contributed to this research. Until today, many examples of carbenes capable of effecting oxidative addition reactions of E–H or E–E bonds (e.g. H–H, N–H, Si–H, P–P) or stabilizing main group element systems with unique reactivities have been reported.3 Key step to these developments however, has been the stabilization of these usually highly reactive species, which have long been regarded as fleeting reaction intermediates and hence mainly been applied in the coordination sphere of transition metals or under extreme reaction conditions. Compared to carbenes, the reactivity of metal carbenoids (metal = s-block metal, Zn) towards E–H and E–E bonds is by far less explored. Since decades, these compounds have been utilized as potent reagents for cyclopropanation reactions.4 However, only recent studies have focused on the stabilization of these usually highly reactive and thermally labile compounds and the development of reactivities beyond classical cyclopropanation reactions.The term carbenoid was first introduced by Closs and Moss in 1964 to describe compounds, “which exhibit reactions qualitatively similar to those of carbenes without necessarily being free divalent carbon species.”5 This distinction to carbenes became necessary to explain the observed selectivity differences in cyclopropanation reactions with carbenes and carbenoids.6 Thereby, the authors noticed that – contrary to diphenylcarbene – the use of the Li/Br carbenoid prepared from methyllithium and diphenyldibromomethane in the cyclopropanation of (Z)-2-butene allows for stereo control (Scheme 1), thus suggesting the presence of an intermediate which is similar, but not equal to a carbene.
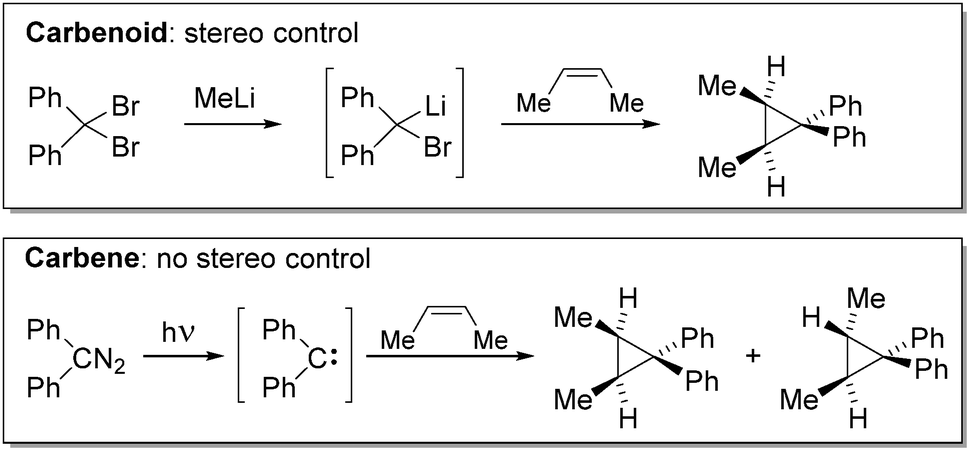 | ||
| Scheme 1 Selectivity differences in cyclopropanation reactions with carbenes and carbenoids. | ||
In general, the carbenoid character was found to be realized in systems with a metalated carbon atom which additionally bears a leaving group X (Fig. 1), i.e. species of the general constitution R2C(X)M (M = s-block metal, Zn). This special constitution of carbenoids results in their ambiphilic character, which becomes evident from their resonance structures (see Fig. 1 for a Li/Cl carbenoid). Historically, cyclopropanations were one of the first reactions that demonstrated this ambiphilicity of carbenoids and their propensity to undergo salt eliminations.4 The fact that metal organyls – despite of their carbanionic nature – exhibit an electrophilic character fascinated chemists ever since. Further reactivities were uncovered demonstrating this electrophilicity, such as C–H activation reactions (mostly intramolecular decomposition reactions) or rearrangements (e.g. the Fritsch–Buttenberg–Wiechell).7 However, the high reactivity and the often observed thermal lability turned out to be the limiting factor to many studies and the development of broader synthetic applications. Hence, until today the most studied reactivity of carbenoids remains the cyclopropanation of olefins.
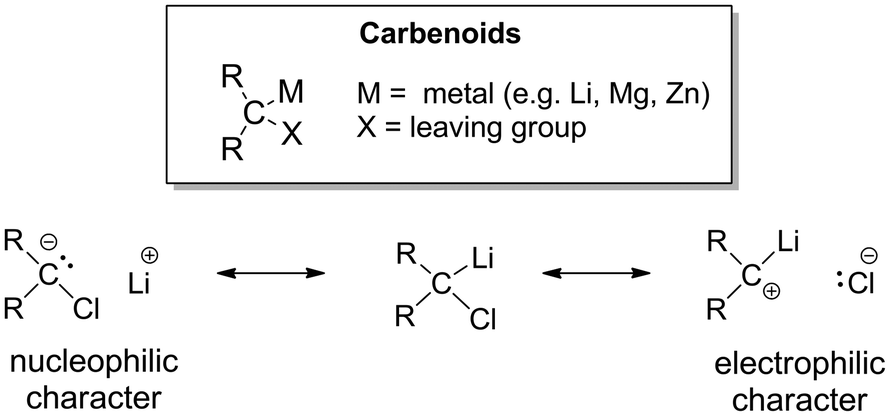 | ||
| Fig. 1 (top) Definition of carbenoids; (bottom) resonance structures of Li/Cl carbenoids. | ||
Recent years however, have experienced a renaissance in carbenoid chemistry. With the first isolation and the stabilization of these compounds novel reactivity patterns were uncovered and many different carbenoid systems – including transition metal species8 – were developed. In this Feature article we highlight recent developments in this chemistry. Thereby, we will focus on s-block metal systems (including Zn carbenoids) with emphasis on the alkali metal compounds. Since the last detailed review on carbenoids published by Boche in 20019 many advances have been made in the stabilization, isolation and structural characterization of these reagents. Hence, we will start with general aspects on carbenoid stability – with special focus on its adjustability – and general preparation methods as well as structural properties. Several reviews on the chemistry of carbenoids, especially on their application in organic synthesis, have appeared over the years.10 Hence, reactivities towards carbon compounds will not be covered here. Instead, we will concentrate on recent developments in carbenoid chemistry towards applications in main group element and transition metal chemistry.
1. The carbenoid character and thermal stability
The fact that metal carbenoids – despite of their carbanionic nature – exhibit an electrophilic character early attracted interest of many synthetic chemists. However, the thermal lability involved with the “uncontrolled” α-elimination of the metal salt and the liberation of an extremely reactive carbene species, limited early investigations of the structural properties and reactivities. Later, X-ray crystallography11 and NMR spectroscopy12 as well by computational methods13 showed that the instability of carbenoids can be rationalized by their unique hybridization compared to the protonated compounds. As such, carbenoids possess a higher p-character in the C–X bond compared to their protonated congeners, which results in the facilitated elimination of X− and MX, respectively. However, there are several factors that influence the stability of a metal carbenoid and thus its carbenoid character (Fig. 2): (a) the M/X combination, (b) the substitution pattern and (c) the solvent and/or presence of additional donor bases or metal salts. By adjustment of these parameters the thermal stability and reactivity of carbenoids can be controlled, thus also allowing the preparation and isolation of systems that are stable at room temperature (vide infra). However, the fact that many parameters affect the carbenoid stability also results in the complexity of these systems. Only under the very same reaction conditions the stability of different carbenoids can be compared to each other. Hence, many examples exist in literature, which describe different stabilities for one and same carbenoid.14 Reactivity and stability control in carbenoid chemistry however, is highly desirable not only to broaden the scope of applications, but also to offer alternatives to synthetic procedures which require the use of expensive transition metals and/or the use of highly reactive or toxic reagents mainly limited to aryl-functionalized systems, such as diazo or ylide compounds.8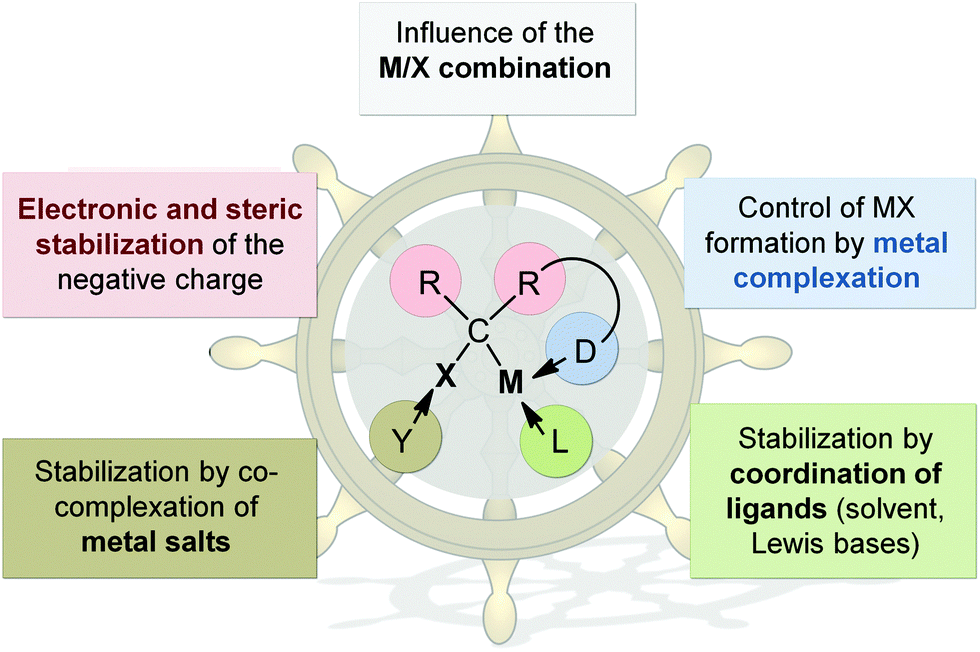 | ||
| Fig. 2 Factors that determine the stability of carbenoids. | ||
The M/X combination
Regarding the nature of the metal, one typically observes an increased stability in the series Li < Mg < Zn. This trend follows the general observations made for simple metal organyls and is due to the decreasing polarity f the M–C bond within this series of metals. This has for example been demonstrated by Boche and co-workers by means of metalated oxazoles (Scheme 2).13 While the zinc compound 1-Zn could be isolated at −30 °C, the lithium compound was found to be unstable even at −78 °C decomposing via Li/OR elimination to the corresponding (Z)-2-isocyanolithium enolate 2-Li. Computational studies also revealed a higher p-character in the C–OR bond in 1-Li compared to 1-Zn and thus confirmed the more pronounced carbenoid character for the lithium system. Very recently, our group reported on stability studies of a series of Li, Na and K carbenoids.15 VT-NMR experiments revealed a higher thermal stability of the heavier congeners, which thus allowed – in contrast to the lithium compounds – their isolation at room temperature. The reason for this increased stabilization is presumably the lower Lewis acidity of the heavier metals, which weakens the M–X interaction in the carbenoid. This effect overcompensates the higher reactivity of the carbanion due to the higher polarity of the M–C bond.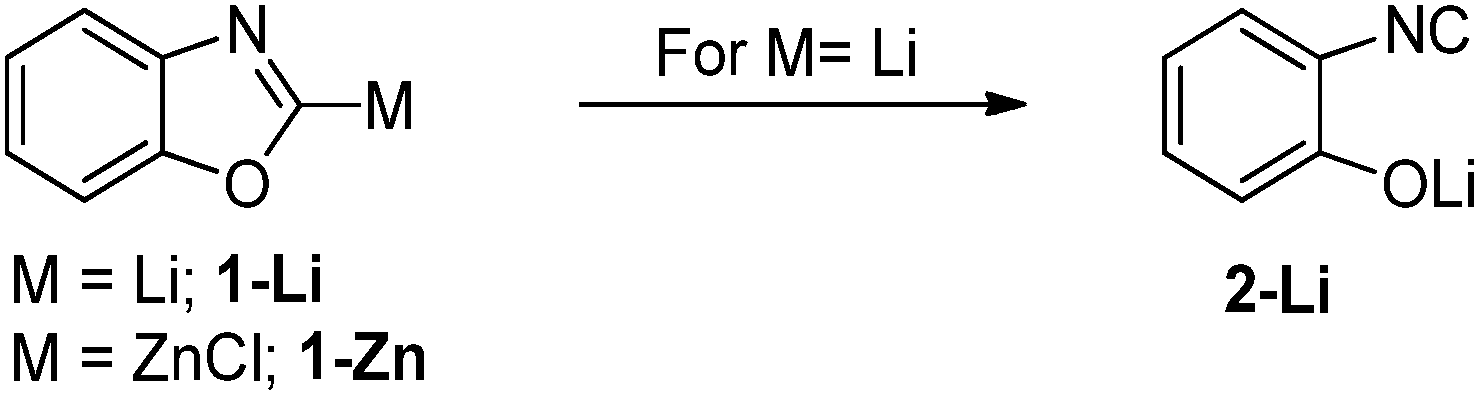 | ||
| Scheme 2 Lithiated and zincated oxazoles 1-M. | ||
Besides the nature of the metal, the leaving group X crucially determines the reactivity and stability of carbenoids. In general, halogens and ethers are the leaving groups leading to the most pronounced carbenoid character. Li/Hal carbenoids are regarded as the most reactive species, which often have to be handled at temperatures below −78 °C. This has already been noted in the late 1960s in seminal studies on simple systems such as Li–CH2Cl or Li–CHBr2, reported by Köbrich and coworkers.14,16 Computational studies on the nucleofugal ability revealed a decreasing carbenoid character in the series LiCH2F > LiCH2Cl > LiCH2Br > LiCH2I.17 In general, the carbenoid character for LiCH2X was found to be more pronounced with groups of the second row of the periodic table (X = NH2, OH, F) than with their heavier congeners (X = PH2, SH, Cl). This was referred to the decreasing electronegativity and the increasing anion-stabilizing ability (via polarization and negative hyperconjugation effects) when going down the group of the periodic table.18 Until today, Li/F systems are regarded as the “beast” in carbenoid chemistry.
In contrast to ethers α-lithiated amines generally possess no carbenoid character.19 One exception are aziridines, which undergo α-elimination reactions due to the strain in the three-membered ring.20 Also lithiated thioethers have shown to undergo MSR elimination reactions, which however strongly depend on the substituent at sulfur. While most thioethers behave like simple organolithium compounds, α-lithiated arylthioethers such as LiC(SAr)321 or PhSCH2Li22 behave like carbenoids.23 Very recently, Steinborn and coworkers also demonstrated that sulfoxides may exhibit a carbenoid character.24
The substitution pattern
As in the case of simple metal organyls the reactivity and stability of the carbanionic center can crucially be influenced by the substitution pattern. In general, electron-withdrawing groups stabilize the carbenoid and thus decrease their tendency for MX elimination. For example, tribromomethyllithium is more stable than the dibromo compound and can even be prepared from dibromomethyllithium and bromoform.25 The right choice of the substitution pattern, even allows for the stabilization of the typically highly reactive Li/Cl systems at room temperature. This has first been demonstrated by Le Floch26 and coworkers in 2007 and confirmed by Gessner,27 Mézailles28 and others.29Fig. 3 depicts a series of Li/Cl carbenoids and their decomposition temperature. The comparison impressively demonstrates the remarkable impact of the substituents and the tunability of carbenoid stability. For example, while dichloromethyllithium (3) is only stable at temperatures below −78 °C,14 substitution of the carbenoid center by thiophosphoryl moieties produces the room temperature stable system 8. It is interesting to note, that the thermal stability is not only determined by the stabilization of the carbenoid carbon center but also by the existence or absence of possible decomposition pathways. Hence, due to intramolecular B–H activation the phosphino borane compound 6 is less stable than the symmetric bis(thiophosphoryl) system 8.28a Besides the introduction of electron-withdrawing substituents the carbenoid carbon center can also be stabilized by a change in hybridization from sp3 to sp2. The higher s-character of the C–M bond results in its stabilization and hence in a generally greater stability of vinylic carbenoids.30 Consequently, the first isolation of a Li/Hal carbenoid has been accomplished by Boche and coworkers with vinyl system 4 (vide infra), which is stable at −78 °C.11 The phosphaalkene 5 reported by Niecke had long been the most stable Li/Cl system known.31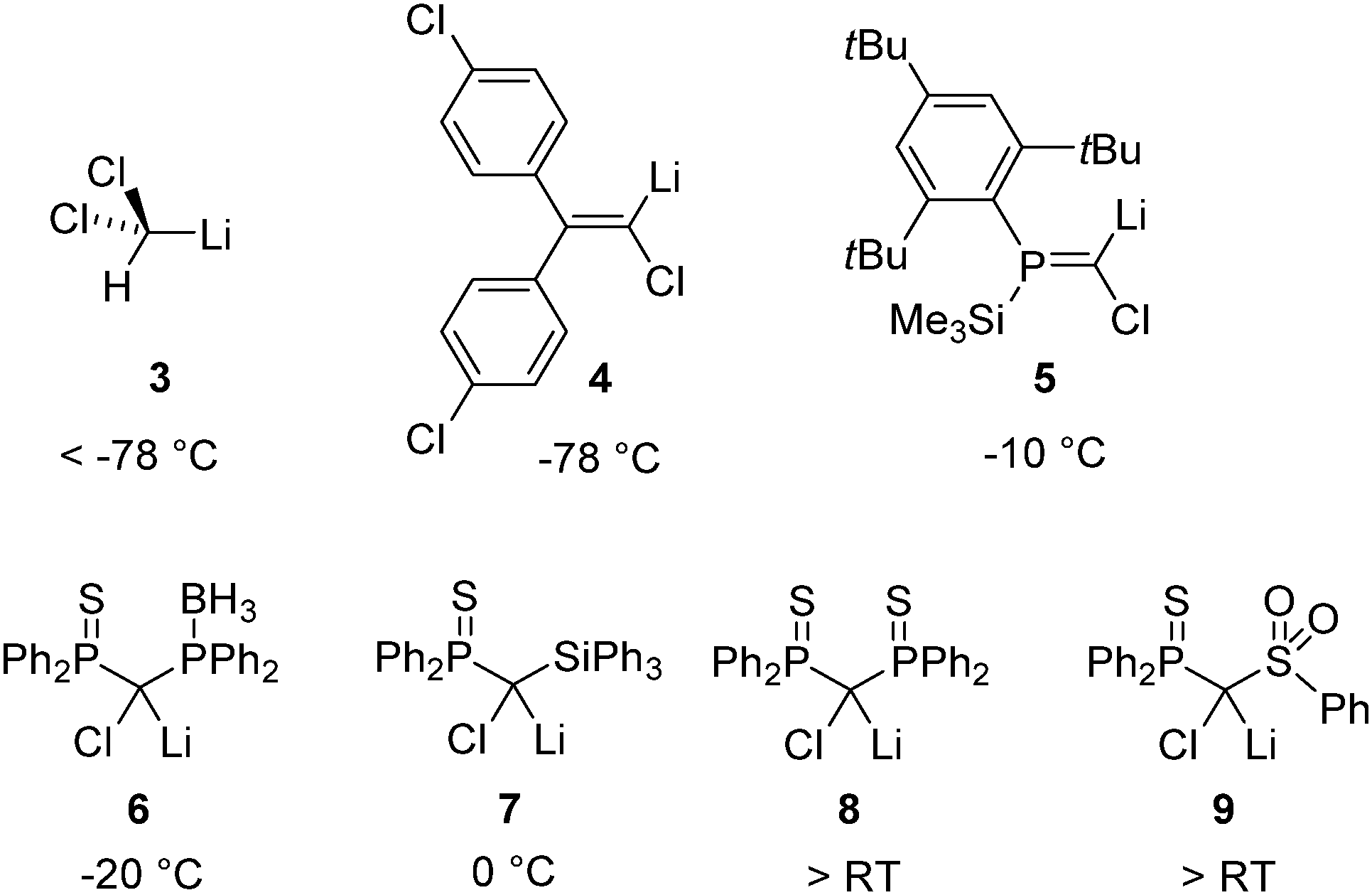 | ||
| Fig. 3 Examples of isolated Li/Cl carbenoids and their decomposition temperature in solution. | ||
Additional donor bases and metal salts
The decomposition of carbenoids by α-elimination can significantly be controlled by complexation of the metal by donor bases. This coordination can occur in an intramolecular fashion in case of donor-functionalized substituents (cf. carbenoids 6–9) or in an intermolecular fashion when using coordinating solvents or additional Lewis bases. Typically, complexation of the metal by donor bases results in a decreased Lewis acidity of the metal, its increased solubility and presumably in many systems in the disruption of the M–X interaction. This considerably hampers metal salt elimination and thus increases the thermal stability. Although systematic studies on the stability of a given carbenoid in different solvents and/or in the presence of different Lewis bases are rare, solvent effects have early been reported. For example, Köbrich et al. noticed in seminal studies on simple lithium carbenoids higher selectivities when reactions were performed in THF relative to those in diethyl ether.32 They also determined critical ratios of Et2O/THF, which still gave selective conversion and suggested the so-called Trapp mixture (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O:petrol ether = 4:1:1) as ideal solvent mixture for the preparation of carbenoids. The employment of strong (often multidentate) N- or O-donor ligands such as TMEDA or PMDETA is also beneficial for the isolation of carbenoids, as has been shown in the case of the Li/Cl systems 3–5 (vide infra), and can also affect the stereochemistry of their transformations.33
:Et2O:petrol ether = 4:1:1) as ideal solvent mixture for the preparation of carbenoids. The employment of strong (often multidentate) N- or O-donor ligands such as TMEDA or PMDETA is also beneficial for the isolation of carbenoids, as has been shown in the case of the Li/Cl systems 3–5 (vide infra), and can also affect the stereochemistry of their transformations.33
Besides influencing carbenoid stability by complexation of the metal by donor bases, the stability can also be affected by coordination to the nucleofugal group. Salt effects in α-elimination reactions have already been noted in the 1960s,34 and were later also confirmed for metal halide carbenoids by Villieras and coworkers, who observed higher yields in reactions of bromomethyllithium, BrCH2Li, when adding one equivalent of LiBr.35 This was confirmed by several groups – particularly Matteson36 and Pace37 – highlighting the effectiveness of using the mixed metalating reagent, MeLi·LiBr, for carbenoid formation. The stabilizing effect of metal salt is assumed to origin from co-aggregation of the carbenoid with LiX, which weakens or disrupts the internal M–X interaction within the carbenoid (Fig. 4).35
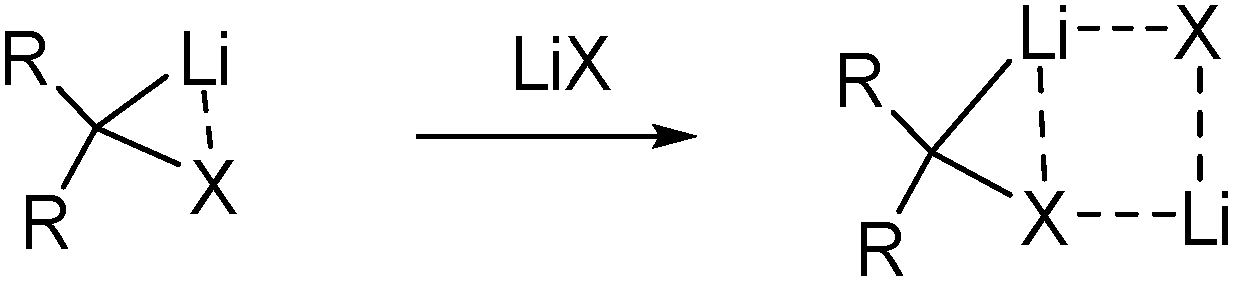 | ||
| Fig. 4 Stabilization of lithium carbenoids by lithium halide complexation. | ||
Overall, thermolability due to MX α-elimination is a typical and characteristic property of carbenoids. This property however, is influenced by many factors, which – on the one hand – makes it difficult to predict stabilities under certain reaction conditions. On the other hand, the versatile adjustability of the stability also allows for the fine-tuning and control of reactivity (vide infra).
2. Preparation of carbenoids
Despite their thermal sensitivity several routes for the preparation of carbenoids have been developed over the years. Typically carbenoid generation is performed at low reaction temperatures. In most of the cases no isolation of the generally highly sensitive compounds has been attempted. Instead they were usually applied in situ shortly after their preparation to prevent decomposition. In most of the cases, the choice of the right reaction conditions (vide supra), above all the solvent, temperature, metalation reagent and/or the presence of additional donor bases turned out to be critical for the successful and selective preparation.The preparation of carbenoids either proceeds via introduction of the metal or the leaving group in the last step of the synthesis. Due to the requirement of geminal dimetalated compounds in the latter case, metalation is the common route to carbenoids. In the majority of reports, this includes the two classical pathways via deprotonation (direct metalation) or halogen–metal exchange (Scheme 3). Particularly bromine– or iodine–metal exchange reactions are widely used for the generation of lithium carbenoids. On the contrary, only a limited number of halogen–magnesium exchange reactions have been reported.38 In case of halomethyllithium compounds often the Barbier method is applied for the generation and reaction of the carbenoid.39 This includes the synthesis of the carbenoid (mostly by Li/I or Li/Br exchange e.g. ICH2Cl + MeLi) already in the presence of the electrophile, which thus ensures immediate reaction of the reactive halomethyllithium species directly after its formation. Although this strategy has successfully been employed for the application of the carbenoid in organic synthesis,40 limitations have been noticed in cases when formation of the carbenoid is slower than the reaction of the substrate with the alkyllithium reagent.41
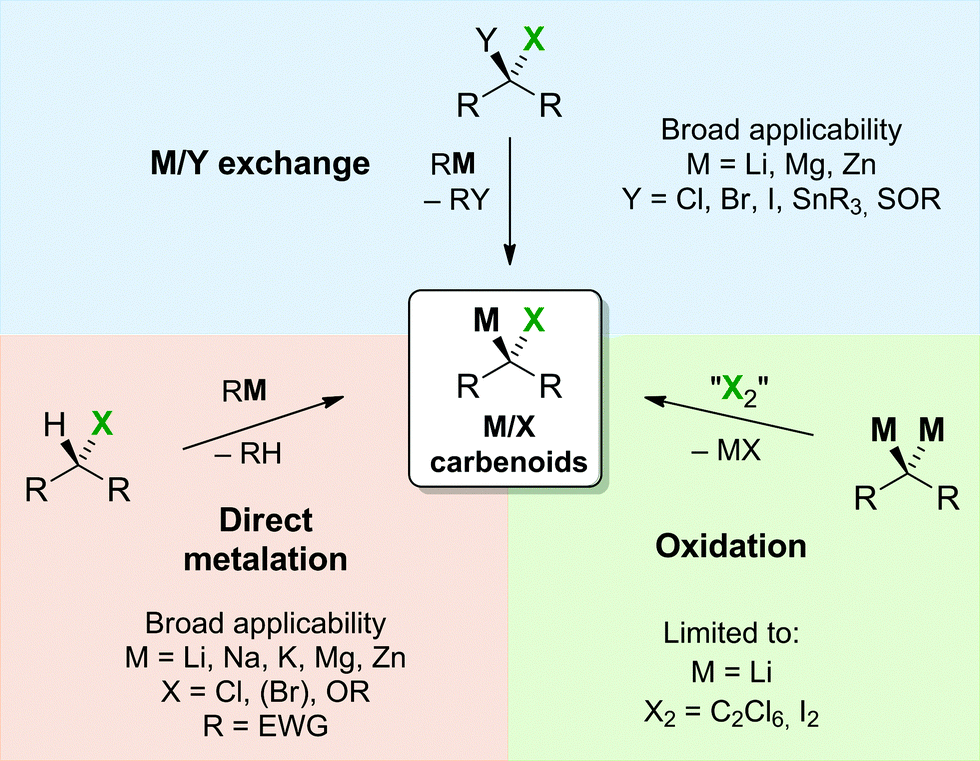 | ||
| Scheme 3 Preparation methods for carbenoids. | ||
Besides the classical pathways via deprotonation or halogen–metal exchange reactions a number of other strategies to access carbenoid species have been reported, yet most of them only applicable to a limited number of substrates. The most successful routes include further exchange reactions using tin compounds or sulfoxides. Tin/lithium exchange for example has only recently been used by Hammerschmidt to access enantiomerically pure chloromethyllithium.14d Sulfoxide/metal exchange was first reported by Satoh and coworkers by means of magnesium carbenoids42 and later transferred to the lithium compounds.43 In case of lithium carbenoids the advances in the preparation of dilithiomethanes resulted in the development of an elegant method, namely the mild oxidation of the dilithium salt. This was first demonstrated by Le Floch with the synthesis of a Li/Cl carbenoid using hexachloroethane as oxidation reagent (R = Ph2P(S) in Scheme 2).26 Later the analogous iodo compound could be accessed by employment of iodine.29 Because of the limited number of readily available methandiide precursors this method remains applicable for only a few systems.27,44
3. Structures and characterization of carbenoids
NMR-spectroscopy
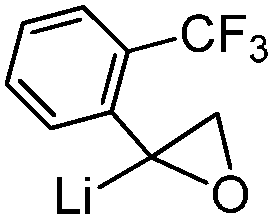
The unique reactivity of carbenoids, particularly compared to simple metal organyls, early raised the question of their electronic and structural properties. Due to the high reactivity, and thermal lability, early studies were restricted to in situ preparation and characterization without isolation or even purification. First informative spectroscopic studies were thus solution NMR experiments, which are mostly connected with the group of Seebach.45 Detailed studies on different lithium carbenoids showed a distinct deshielding of the carbenoid carbon atom with a down-field shift relative to the protonated analogue of up to ΔδC = 280 ppm in the 13C NMR spectrum. For example, lithation of chloroform to trichloromethyllithium resulted in a down-field shift by ΔδC = 65.9 ppm, lithation of bromoform to tribromomethyllithium in a shift by ΔδC = 142.5 ppm (Table 1).45b This observation contrasts the typical high-field shift for simple organolithium compounds,46 but compares well with the NMR spectroscopic properties of free carbenes.47 The 13C NMR shifts and the typically observed deshielding of the carbenoid carbon atom could also be reproduced by IGLO (individual gauge for localized orbitals) calculations. Origin of the deshielding is not the partial charge at the carbon atom – which is often similar to that of the protonated congener – but the energy of the bonding σ(CLi)- and the antibonding σ*(CX) orbitals. The higher the σ(CLi) and the lower the σ*(CX) orbital, the greater the deshielding.| Entry | Carbenoid | δ(13C) [ppm] | Δδ(13C) Li/H [ppm] | Ref. |
|---|---|---|---|---|
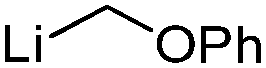
| 1 | LiCH2Cl | 57.9 | 32.3 | 48 |
| 2 | LiCHCl2 | 105.5 | 50.0 | 39 |
| 3 | LiCCl3 | 145.9 | 65.9 | 39 |
| 4 |
|
74.5 | 19.7 | 55 |
| 5 |

|
92.2 | 29.9 | 55 |
| 6 |
|
≈83.3 | ≈31.3 | 49 |
| 7 |

|
M = Li: 37.6 | M = Li: −4.7 | 15 |
| M = Na: 42.9 | M = Na: 0.6 | |||
| M = K: 42.6 | M = K: 0.3 | |||
| 8 |
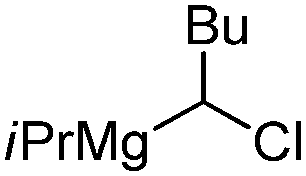
|
68.8 | 24.8 | 50 |
| 9 |
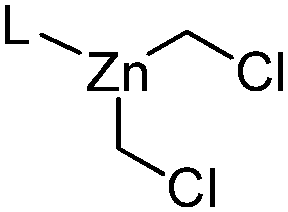
|
L = DME: 29.6 | L = DME: 4.0 | 51 and 52 |
| L = Bipy: 32.9 | L = Bipy: 7.3 | |||
| 10 | XZnCH2I | X = (BuO)2P(O)O: −23.7 | X = (BuO)2P(O)O: −0.1 | 53 and 54 |
| X = RO ≈−27.0 | X = RO ≈−3.4 |
The deshielding of the carbenoid carbon atom was also observed for Li/OR carbenoids, albeit less pronounced than in case of Li/Hal systems. As such, shift changes of only ΔδC = 7–40 ppm have been reported (cf. entries 4–6, Table 1).55 The weaker deshielding for Li/OR compared to Li/Hal carbenoids is well in line with the generally stronger carbenoid character of the halide systems. However, similarly to the stability of carbenoids, the NMR shift changes are also dramatically influenced by the complexation of the metal or the substitution pattern. The room temperature stable Li/Cl carbenoids 8 and 9 for example showed no deshielding, although exhibiting reactions with LiCl elimination. Likewise, disruption of the C–M bond by strongly coordinating donor bases (e.g. PMDETA or crown ether) leads to smaller shifts than found for structures with a direct metal carbon linkage.55 Hence, the deshielding of the carbenoid carbon atom is indicative for the carbenoid character, but no exclusion criterion. Compared to lithium carbenoids, this property is much less pronounce for zinc and magnesium system. Above all the zinc carbenoids often showed no distinct downfield shift relative to their protonated analogue (see entries 9 and 10).50–54
Besides the 13C NMR chemical shifts also the J(13C,6Li) coupling constant has found to be indicative for a carbenoid character. Typically a large 1J(13C,6Li) value of around 17 Hz is observed due to the higher s-character of the carbenoid carbon atom compared with simple organolithium reagents, which usually show coupling constants between 8 and 10 Hz.48 Accordingly, smaller 1J-coupling constants are found to the other substitutents. For example, lithiation of dichloromethane (1JCH = 178 Hz) to LiCHCl2 results in a by 68 Hz smaller 1JCH coupling constant.48
Computational studies
The molecular structures of carbenoids were first studied by computational methods.56 Seminal studies by the Schleyer group focused on simple systems such as LiCH2X and LiCHX2.57 They revealed a preference of a bridged structure such as I (Fig. 5) with contacts between lithium and both, the carbon atom and the nucleofugal group X. This bridged structure was found to be favored over a classical structure II and carbene donor III and acceptor IV complexes. Although these calculations only considered monomeric structures and no involvement of solvent molecules, experimental structure elucidations later confirmed the existence of all structural motifs I–IV (vide infra). The energetically most disfavoured carbene–acceptor complex has so far only been observed with NHCs.58 Aggregation and Lewis base coordination was later also considered by computational methods, confirming their importance for the structure formation and for mirroring experimental observations.59 Analogous geometries to those reported for halomethyllithium carbenoids were also observed for vinylcarbenoids, also considering solvent effects.60 Overall, structure formation of carbenoids is a rather complex subject. Even the metal salt MX formed upon decomposition of the carbenoid can influence the structure formation. However, such mixed aggregates with lithium halides and lithium methoxide have only poorly been studied computationally as well as experimentally.61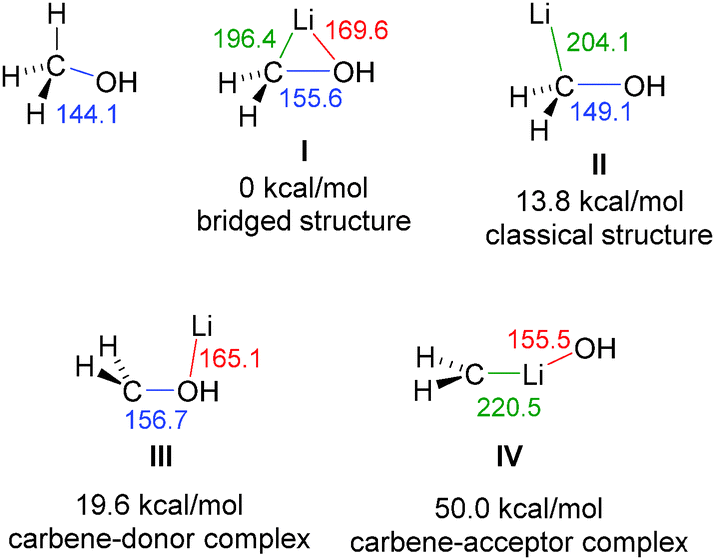 | ||
| Fig. 5 Calculated structures of model carbenoid LiCH2OH and their corresponding energies. | ||
Besides identifying different structural motifs, the computational studies revealed also the elongation of the C–X bond as characteristic structural feature of carbenoids. This elongation is well in line with the electrophilicity and the deshielding of the carbenoid carbon atom found in the NMR experiments. The extent of the elongation however, strongly depends on the M/X combination and the structural motif formed. This is shown in Fig. 5 for the hypothetical carbenoid LiCH2OH, in which the C–O bond elongates by 5–16 ppm relative to methanol depending on the structure. In line with the general reactivities and the carbenoid behaviour, the bond elongation was found to be most pronounced for Li/Hal carbenoids and less significant in the magnesium and zinc compounds.62 However, no direct correlation between the carbenoid character and the C–X bond elongation exists. There are even systems known, which exhibit no C–X bond shortening, despite showing an ambiphilic nature.26–29 For example, thioethers generally show a shortening of the C–S bond upon lithiation, although several examples are known that show carbenoid bahaviour.21,22
DFT studies were also used to elucidate the electronic structure of carbenoids. Thereby, it was found that carbenoids possess a higher p-character in the C–X bond relative to their protonated congeners owing to the higher s-character in the metal carbon bond. This was first demonstrated by calculations on lithium carbenoids17 but also confirmed for zinc and magnesium systems.62 One exception to that rule is the room temperature stable Li/Cl carbenoid 8 (Fig. 3) reported by Le Floch and coworkers.26 Here, no significantly higher p-character was observed in the C–Cl bond.
X-Ray diffraction analyses
 | ||
| Fig. 6 Isolated and structurally characterized Li/OR-carbenoids. | ||
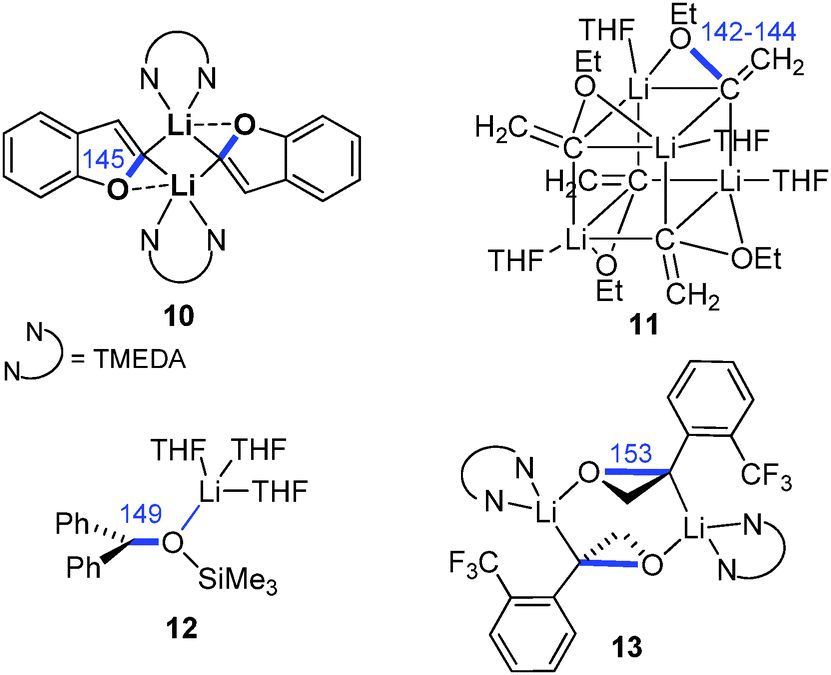
Owing to their extreme thermal lability, the number of isolated and structurally characterized Li/Cl carbenoids has long been limited to compounds 3–5 (Fig. 7).11,26,31 This changed in 2007 with the report of the first room temperature stable Li/Cl system 8 by Le Floch and coworkers. Since then a number of compounds with similar stabilities have been isolated and structurally characterized, all making use of strongly stabilizing substituents. Notably, none of the molecular structures reported so far exhibited the bridged structural motif which was predicted by computational studies and also confirmed in case of Li/OR carbenoids (Fig. 6). Most structures showed either a classical structural motif with a C–Li interaction (cf.3–5) or exclusive coordination of the lithium by additional donor functions in the carbenoid or solvent molecules (e.g.8 and 14/15). Owing to the high reactivity and sensitivity of Li/Hal carbenoids, sufficient complexation of the metal is necessary to prevent or hamper uncontrolled α-elimination. The molecular structures generally showed the expected lengthening of the C–Cl bond. This is particularly true for the thermally labile systems, while the room temperature stable compounds showed no or only slight bond length changes. For example the pyridine adduct of LiCHCl2 (3) exhibited a C–Cl bond elongation of approx. 10 ppm, whereas the bis(thiophosphoryl) system 8 and its derivatives 6, 14 and 15 showed no change relative to the protonated congener.26,28 Interestingly, the iodo congener of 8 was found to be less stable than the chloro compound and decomposed via LiI elimination at room temperature.29 However, the molecular structure also showed no distinct C–I bond elongation, thus demonstrating that the C–X bond lengthening does not necessarily correlate with the carbenoid stability. This is particularly true, when metalation is also accompanied by changes in the hybridization at the carbenoid carbon atom, which – in addition to the structural motif formed – affects bond lengths and angles. The only solvent-separated ion-pair structure of a lithium carbenoid was only recently reported with the crown-ether coordinated compound 7.15 Despite the missing C–Li interaction a slight C–Cl bond elongation was observed. Most remarkably, the crown-ether complex showed a by 20 °C higher thermal stability than its THF analogue (decomposition at 0 °C).
 | ||
| Fig. 7 Isolated and structurally characterized Li/Cl-carbenoids (C–Cl bond lengths are given in blue [pm]). | ||
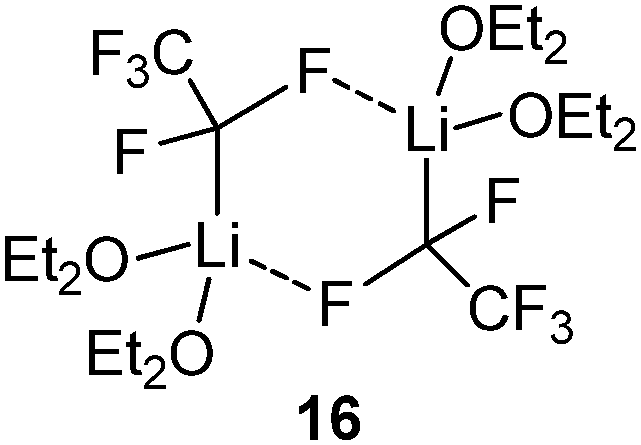
Besides the iodo congener of 8 no other Li/I carbenoid has been isolated and structurally characterized until today. Similarly, the structures of Li/Br and Li/F carbenoids are also much less explored than their chloro analogues. The only structure of a Li/Br system has been reported by Niecke and coworkers with the bromo congener of 5.68 The analogous Li/F system69 showed an extraordinary stability even at −50 °C, which thus facilitated its isolation. Here, the stability was not limited by α-elimination of LiF, but by solvent degradation and protonation of the carbenoid. In the molecular structure, the Li/Br and Li/F systems formed monomers analogous to 5 with no contact between the lithium and the halide. This is particularly noteworthy in case of the fluoro compound, because of the usually stronger Li–F interactions. The bromo as well as the fluoro carbenoids showed elongation of the C–X bonds, which was more pronounced in the Li/F (7.0%) than in the Li/Br (4.5%) system. The first sp3-hybridized Li/F carbenoid was recently reported by Hoge, Mitzel and coworkers.70 They succeeded in the isolation of the highly reactive F3CCF2Li as its diethyl ether adduct 16. LiC2F5 is potentially explosive and has to be handled below −60 °C to exclude decomposition reactions.7116 forms a dimeric structure in the solid state, which is dominated by Li–F interactions. Accordingly, the C–F bonds at the carbenoid carbon atom are elongated by 7 and 12 ppm compared to the protonated congener.
The first sodium and potassium carbenoids have only recently been reported by our group.15 Interestingly, Na/Cl carbenoid 17 and its potassium analogue 18 showed higher stabilities than their lithium counterpart (Fig. 8). Both formed carbene–donor complex-like structures in the solid state with a contact between the metal and the chloride, but not C–M interaction. While the sodium compound formed a monomeric complex, 18 exhibited a dimeric structure with additional metal arene interactions. In both structures, the C–Cl bond was found to be slightly elongated (Δd = 3 and 5 pm).
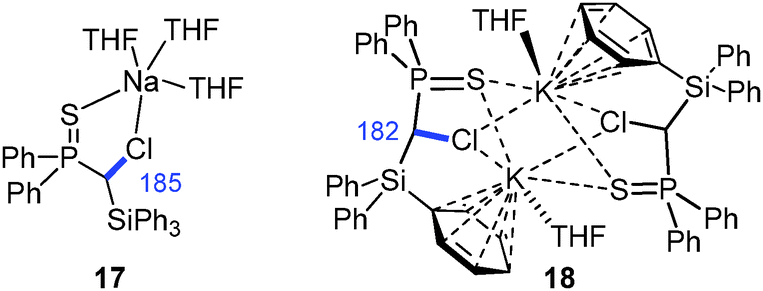 | ||
| Fig. 8 Isolated sodium and potassium carbenoids (C–Cl bond lengths are given in blue [pm]). | ||
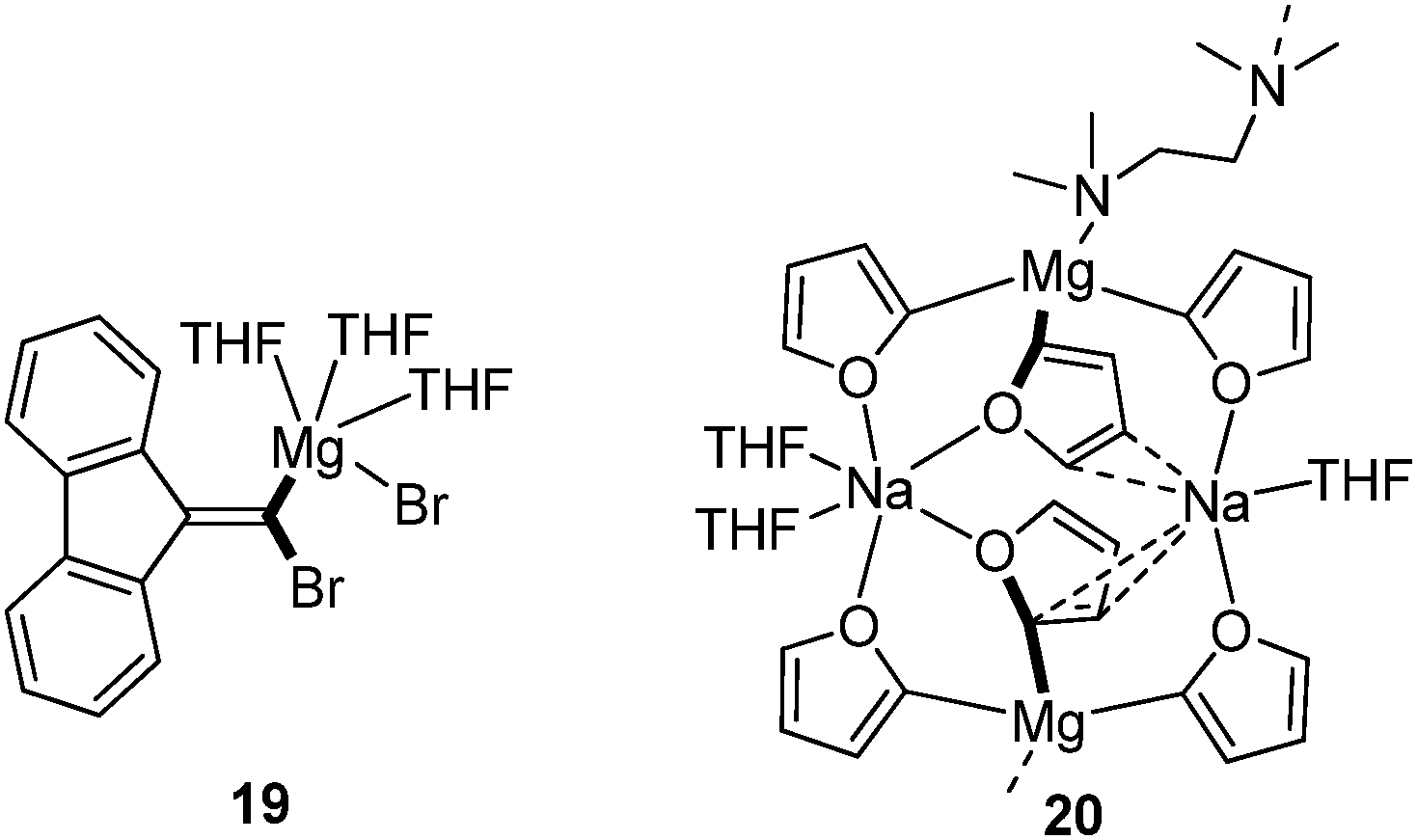 | ||
| Fig. 9 Isolated and structurally characterized Mg carbenoids. | ||
Zinc carbenoids possess a considerably higher stability than their lithium congeners (vide supra). Accordingly, a number of zinc carbenoids, including simple halomethylzinc systems, have been isolated and the structures of several Zn/Hal, Zn/OR and Zn/SR systems have been reported until today, although the carbenoid character of many systems hasn't been explored.73 The class of (iodomethyl)zinc carbenoids are – without doubt – the most important and most frequently applied zinc carbenoids. Depending on the substitution pattern at zinc, these systems are typically classified as Simmons–Smith (IZnCH2I),4 Furukawa (EtZnCH2I),74 Wittig–Denmark (ICH2ZnCH2I)75 or Charette-type (ROZnCH2I)54 reagents (Fig. 10a). Until today, nine (iodomethyl)zinc carbenoids have been characterized by X-ray diffraction analysis,76 while, no structure of a Furukawa-type reagent has been reported. The first structure elucidation of a zinc carbenoid was accomplished by Denmark and coworkers in 1991 with the bis(iodomethyl)zinc complex 21.77 Coordinated by a bis(ether) carbenoid 21 formed a monomeric structure in the solid state. Although no deshielding of the carbenoid carbon atom was observed, the C–I bond lengths were longer than those later determined in other (iodomethyl)zinc compounds such as 22 or 23 (Fig. 10b).78 The phosphate system 22 and derivatives thereof are particularly interesting, since they were found to be highly stable systems that can be stored for days at −22 °C, while keeping their reactivity in cyclopropanation reactions.78b A remarkable Zn/OR system was reported by Mulvey and coworkes with compound 24, which was obtained by direct metalation of THF using a mixed metal base.79 While the use of lithium bases typically results in the degradation of THF after its metalation 24 could be isolated at room temperature and was used for further functionalization reactions.
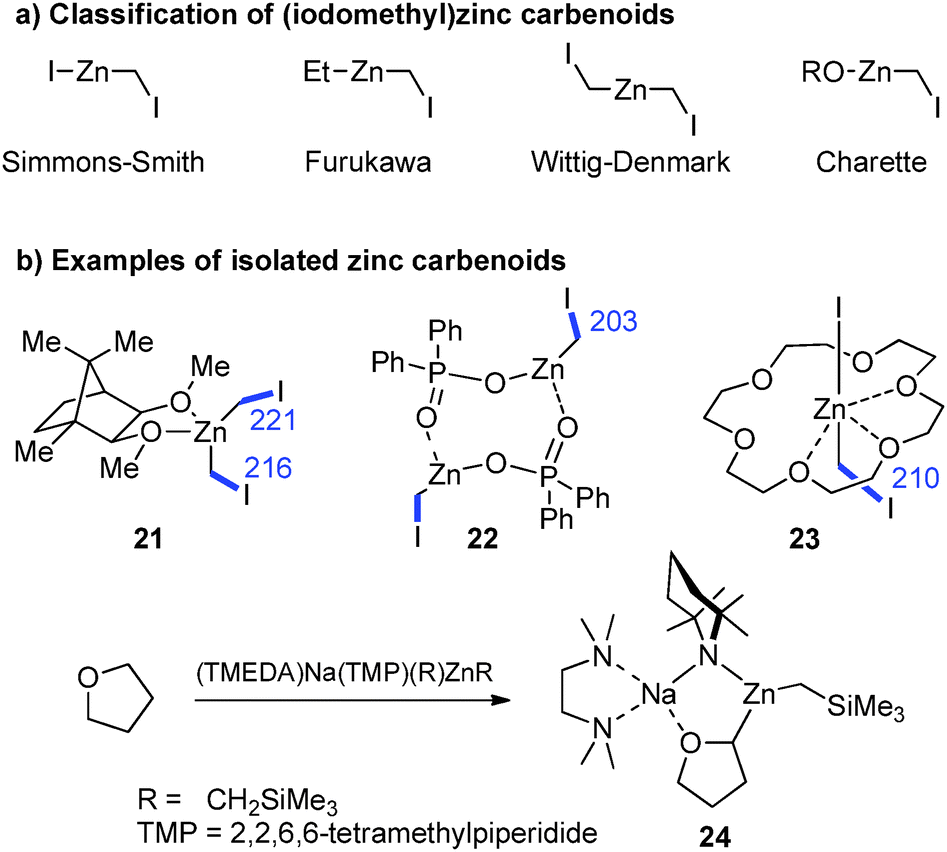 | ||
| Fig. 10 Isolated and structurally characterised Mg and Zn carbenoids. | ||
4. Reactivity of carbenoids towards main group element compounds
Boron chemistry
While carbenoids are widely applied in organic synthesis, their reactivity towards main group element compounds and transition metals is much less explored. In the case of main group element chemistry boron compounds are the by far most intensively studied compounds in carbenoid chemistry. One of the earliest examples was reported by Köbrich in the 1960s. He observed the homologation of triphenylborane via a 1,2-metalate rearrangement in the intermediate ate-complex 26-Int when treated with different Li/Cl carbenoids (Scheme 4).80 However, the real potential of this transformation, particularly in organic synthesis, has only recently been discovered. This was particularly connected with the observation that also boronates40c – as well as diboranes and silylboranes81 – can be used in this reaction providing starting point for further functionalizations. In the past years Aggarwal, Blakemore and others impressively extended this lithiation–borylation reaction, thus establishing stereoselective procedures as well as iterative pathways to build-up long alkyl chains.82 Li/Cl carbenoids as well as lithium carbamates and benzoates were employed in this chemistry, whereat the latter showed higher stabilities and have thus more successfully been employed until today.83,84 The broad utility of the reaction has led to its application in total synthesis.85 A particularly impressive example reported by Aggarwal and coworkers is shown in Scheme 4. Iterative lithiation–borylation–protodeboronation allowed the coupling of smaller fragments such as 27 and carbenoid 28 to finally build up hydroxyphthioceranic acid 29.82a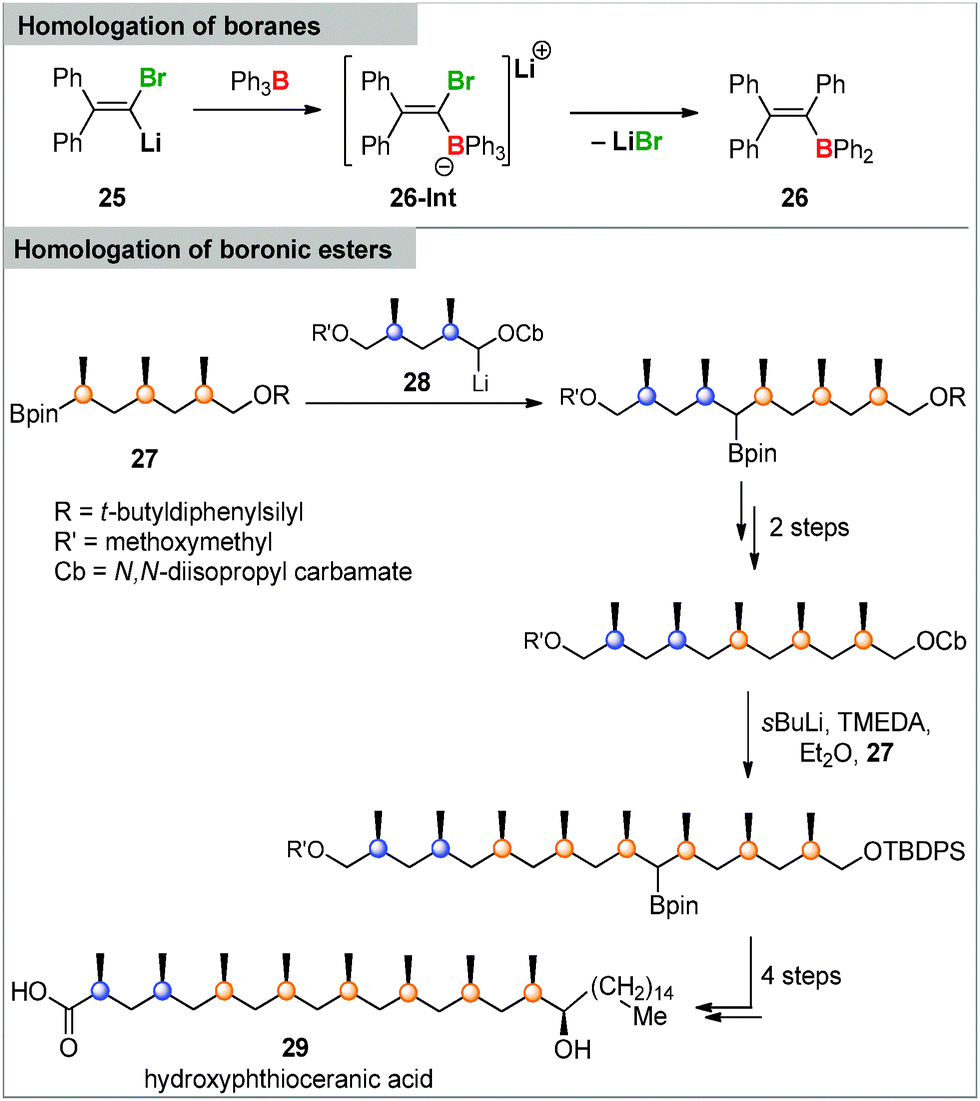 | ||
| Scheme 4 Triphenylborane homologation and total synthesis of hydroxyphthioceranic acid 29via a lithiation–borylation–protodeboronation sequence. | ||
Besides supporting the homologation of triarylboranes and boronates carbenoids were also found to insert into the B–H bond of simple BH3 Lewis base adducts. This was first reported in 2013 by Mézailles, So and coworkers by means of the phosphino-borane functionalized Li/Cl system 6 (Scheme 5).28a This carbenoid – contrary to the bis(thiophosphoryl) system 8 – was found to be unstable at room temperature undergoing an intramolecular B–H bond activation at temperatures above −20 °C. The reaction lead to the elimination of LiCl and the formation of the corresponding borane 30 stabilized through intramolecular coordination via the thiophosphoryl moiety. This reactivity was extended to an intermolecular activation reaction using carbenoid 8 and BH3·THF. Interestingly, B–H bond activation with the more reactive Li/Cl silyl substituted system 7 did not result in an analogous borane formation but selectively delivered borate 31.86 The reaction was found to require a slight excess of borane and was accompanied by chloroborane formation.
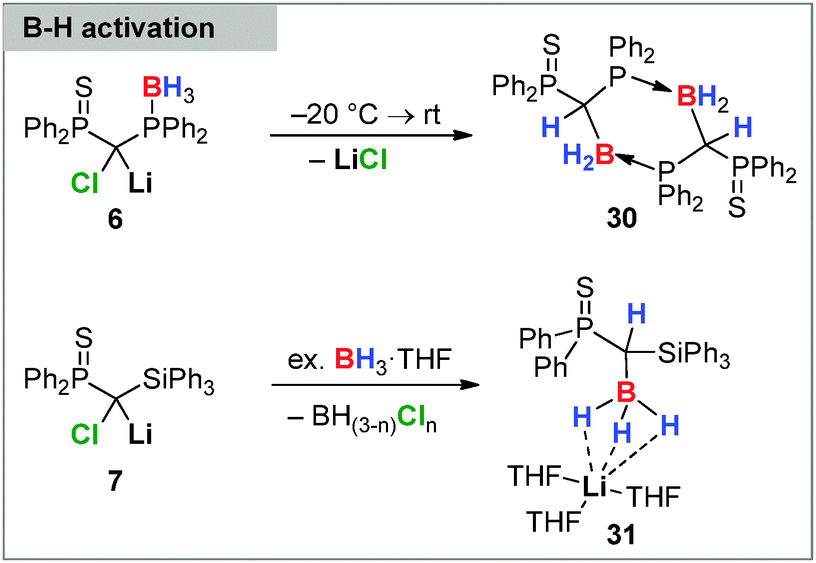 | ||
| Scheme 5 B–H activation with Li/Cl carbenoids. | ||
Despite the different products formed the B–H bond activation reactions were found to proceed via an analogous step-wise mechanism including nucleophilic attack of the carbenoid at the borane followed by H/Cl exchange. The activation barrier of the nucleophilic attack with replacement of the corresponding Lewis base was found to determine the viability of the processes. As such, more stable Lewis base adducts, such as BH3·NEt3, were found to be unreactive due to the elevated activation barrier.
The potential of carbenoids in catalytic transformations was only recently demonstrated by Mézailles and coworkers by means of the reduction of CO2 with BH3.28b Starting point of these studies was the B–H activation with the bis(iminophosphoryl) substituted carbenoid 32, resulting in the formation of compound 33, in which a BH2+ group is stabilized by the two PN moieties (Scheme 6). Both, the carbenoid 32 as well as the activation product 33 were successfully used as catalyst in the CO2 reduction to selectively deliver methanol derivatives in up to quantitative yields and TONs of up to 2646.
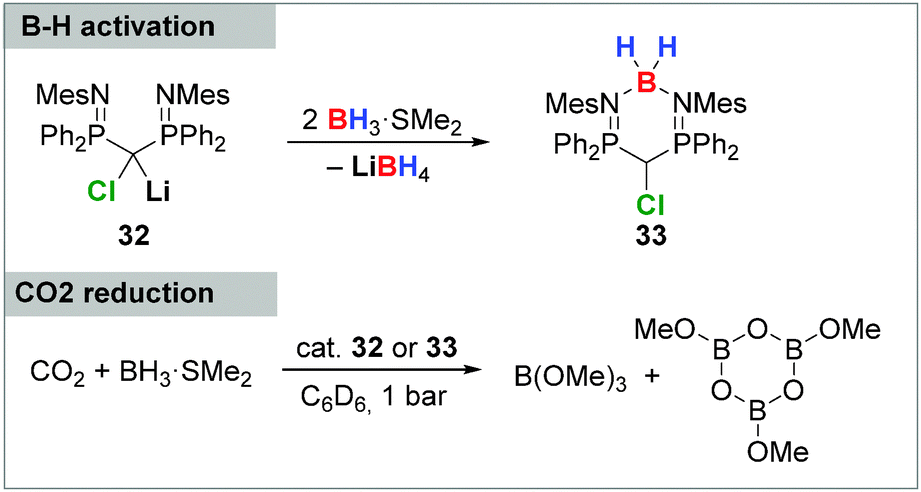 | ||
| Scheme 6 B–H bond activation with carbenoid 32 and its application in CO2 reduction. | ||
Phosphorus chemistry
Applications of carbenoids in phosphorus chemistry have only scarcely been explored. Early reports by Märkl focussed on the reactivity towards phosphabenzenes. Thereby, 2,4,6-triphenylphosphabenzene was found to react with LiCHCl2 to benzene derivatives via phosphorus elimination.87 The reaction proceeds at −80 °C via the anionic intermediate 34-Int, which was identified by in situ trapping reactions, but quickly “decomposes” to 34 upon warming to room temperature (Scheme 7).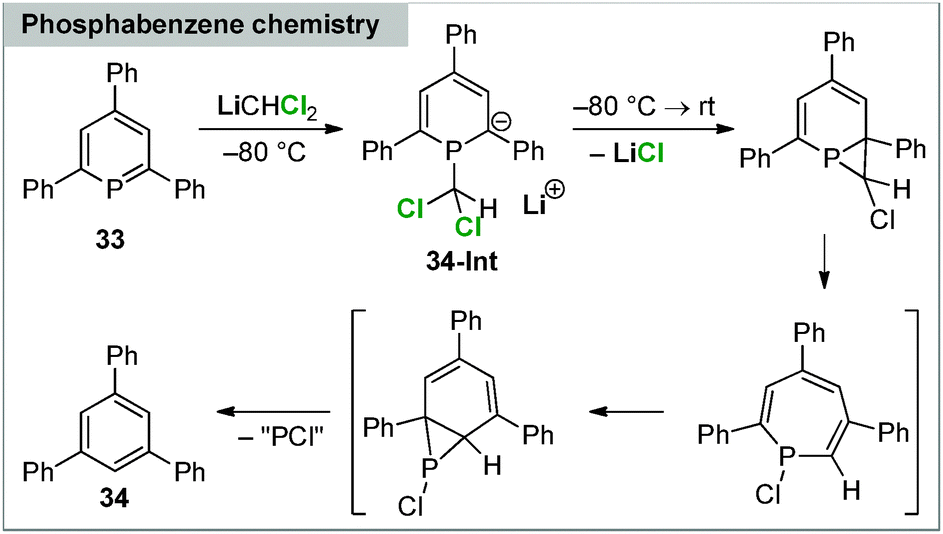 | ||
| Scheme 7 Reaction of dichloromethyllithium with phosphabenzene 33. | ||
A surprising reactivity of electronically stabilized carbenoids was only recently discovered in our laboratories. Treatment of the stabilized Li/Cl carbenoids 7 and 9 (as well as the potassium and sodium derivatives of 7) with secondary phosphines did not result in a P–H activation or homologation analogous to boranes, but in the selective formation of diphosphines (Scheme 8).88 Such a carbenoid-mediated dehydrocoupling reaction has never been observed before with any other substrate. The reaction protocol was transferrable to a series of functionalized phosphines, including chloro-substituents, and provided the diphosphines in high yields. The selectivity of this reaction was found to strongly depend on the stability of the carbenoids. Only the highly stabilized systems gave way to selective dehydrocoupling, while the more labile carbenoids, e.g. LiCHCl2, provided complex product mixtures. This was explained by the mechanism, which first proceeds via deprotonation and phosphide formation. This step is prefered for the stabilized carbenoids, whereas LiCl elimination is competing in case of the labile systems, thus showing that electronic manipulation may give rise to new applications in carbenoid chemistry.
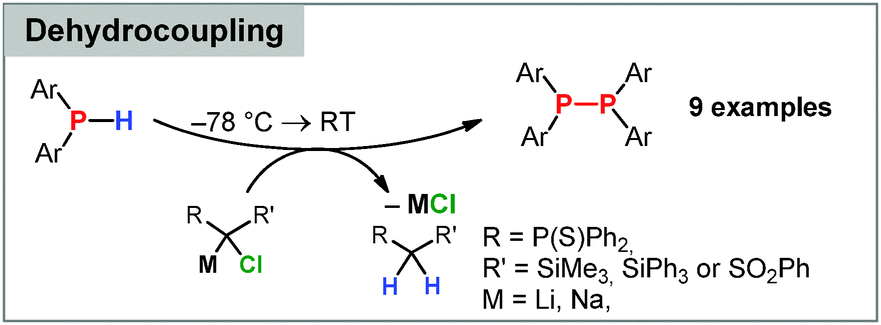 | ||
| Scheme 8 Carbenoid-mediated dehydrocoupling of secondary phosphines. | ||
Sulfur and selenium chemistry
Group 16 element compounds have only recently been employed in carbenoid chemistry. Pace and coworkers observed that lithium carbenoids effectively insert into the S–S bond of disulfides.89 Different Li/X combinations were tested with bromo derivatives giving the best results. The protocol allowed the homologation of a series of functionalized disulfides thus providing a valuable tool for the formation of dithioacetales (Scheme 9). Most remarkably, the same reaction conditions were applicable for the insertion of the carbenoid into the Se–Se bond in diselenides, thus underlining the synthetic benefit of this reaction.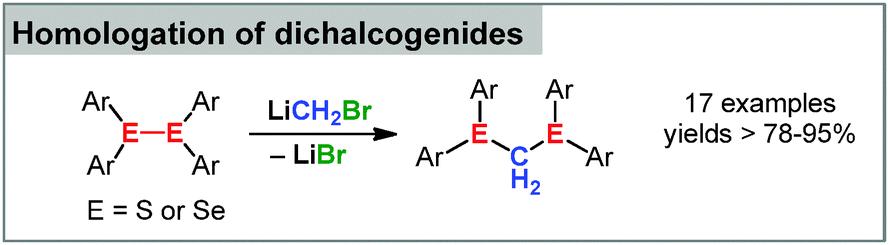 | ||
| Scheme 9 Homologation of disulfides and diselenides. | ||
5. Reactivity of carbenoids towards transition metal compounds
Despite their carbene-like reactivity carbenoids have only scarcely been employed in the synthesis of carbene complexes. This can probably also be referred to the fact that the reaction of carbenoids with transition metal precursors does not necessarily result in the formation of carbene species. For example, the reaction of carbenoids with organo zirconocenes such as 35 results in an insertion of the carbenoid into the Zr–C bond. Protic work-up thus gives way to the corresponding C–C bond formation product (e.g.36, Scheme 10a). This has first been demonstrated by means of acyclic systems90 and was subsequently transferred to zirconacycles, resulting in ring expansion reactions.91 The reactions presumably proceed via nucleophilic attack of the carbenoid to an ate-complex, which undergoes 1,2-rearrangement to the final product (Scheme 10b). An analogous reactivity has also been reported with other metals,90a yet in a less selective manner than was observed for the zirconium compounds, which also proved to be applicable in total synthesis.92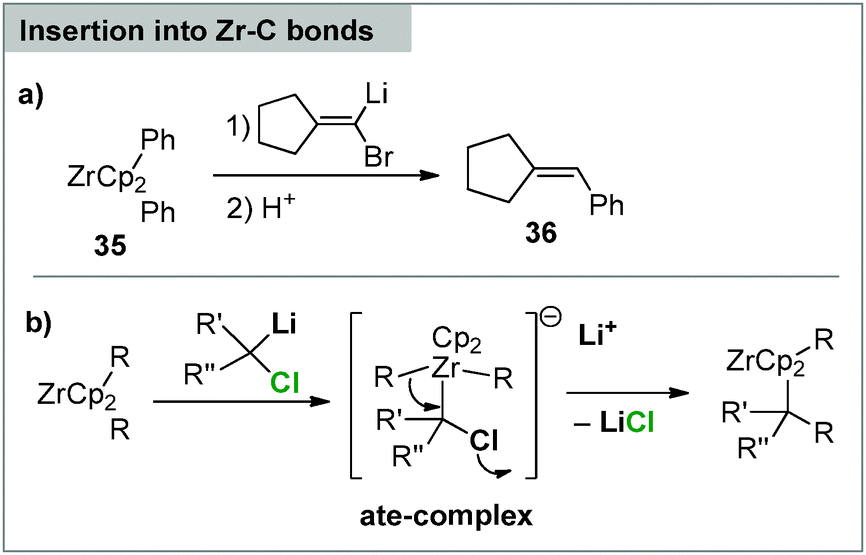 | ||
| Scheme 10 (a) Reaction of lithium carbenoids with organozirconium reagents and (b) the proposed mechanism. | ||
The use of carbenoids as carbene-transfer reagents for the synthesis of carbene complexes has only been demonstrated at a handful of selected examples. One of the first examples focused on the carbocyclic carbene species 39 (Scheme 11), which provides a valuable alternative route to the reaction with a diazo precursor.93 In 2007 Milstein and coworkers reported the first application of a zinc carbenoid in carbene complex formation. Treatment of [Zn(CH2I)2] or [Zn(CHPhCl)2] with the ruthenium(II) precursor [(PPh3)3RuCl2] delivered the corresponding carbene complexes such as 40, including the Grubbs catalyst.94 Mechanistically, the authors suggested a concerted pathway similar to the methylene transfer in cyclopropanation reactions. In contrast, a step-wise mechanism was postulated by Le Floch and coworkers for the formation of the nucleophilic palladium carbene complex 41 from the corresponding room-temperature stable Li/Cl carbenoid 8. The formation of 41 is remarkable, since nucleophilic carbene complexes of late transition metals are still difficult to synthesize via alternative routes. However, our group also experienced limitations to this apparently straight-forward carbene transfer reaction. Employment of the more reactive silyl carbenoids 7 and 42 gave mixtures of the carbene species 43 and thioketone complex 44 formed via sulfur transfer from phosphorus to carbon. This reaction however, turned out to nicely reflect a stability reactivity relationship of carbenoids. As such, the selectivity of palladium carbene complex formation was found to strongly depend on the stability of the carbenoid.95 While the SiMe3 substituted Li/Cl carbenoid 42 selectively delivered thioketone complex 44, introduction of the SiPh3 moiety in 7 – leading to increased carbenoid stabilization through negative hyperconjugation effects – resulted in a 50% conversion to 43. Most remarkably, replacement of lithium by the heavier congeners, Na and K, provided access to the stable carbenoids 17 and 18, which allowed selective carbene complex formation. The examples in Scheme 11 demonstrate that carbenoids can serve as valuable transfer reagents for carbene complex formation. The scope of this reactivity is still by far underexplored. Yet, the recent developments suggest that manipulation of the carbenoid stability may present a useful tool to control the transfer reaction and thus establish carbenoids as viable precursors for carbene complexes.
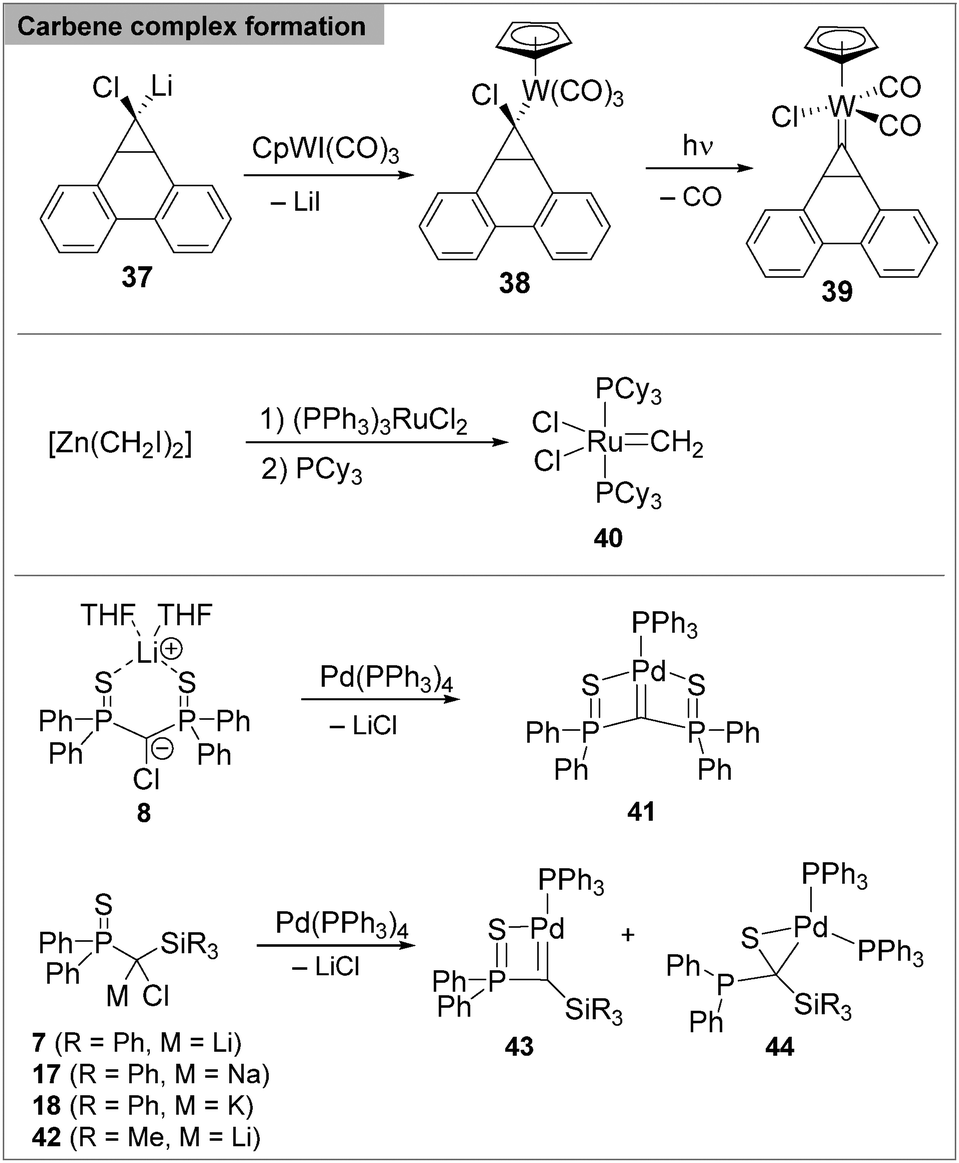 | ||
| Scheme 11 Carbenoids as carbene transfer reagents for the synthesis of transition metal carbene complexes. | ||
6. Conclusions
Carbenoid chemistry has quite a long history, beginning with seminal studies on cyclopropanation reactions, which were followed by a myriad of organic transformations. However, the last two decades have experienced a renaissance in their application with the development of transformations beyond classical organic syntheses. On the one hand this can be ascribed to the nowadays convenient handling of these often highly reactive and thermally sensitive species by standard inert gas techniques. On the other hand, fundamental studies have provided a profound understanding of the electronic structure and aggregation of these species which now allows their controlled stabilization and electronic tuning. This has paved the way for the isolation of carbenoids and their stoichiometric applications as well-defined complexes, which facilitated the development of new reactivity patterns also in main group element and transition metal chemistry. As such, coupling reactions as well as carbene transfer reactions and even catalytic transformations are now part of the repertoire of carbenoid reactivity. With many discoveries only made in the last couple of years the revelation of further applications can be expected, which will further underline the unique reactivity and behaviour of metal carbenoids and their role as extraordinary organometallic reagents.Acknowledgements
This work was supported by the German Science Foundation (Emmy Noether Grant, DA1402/01) and the Fonds der Chemischen Industrie. VHG thanks Prof. Holger Braunschweig and the Institute of Inorganic Chemistry of the University of Würzburg for support and excellent working conditions over the past years and the Ruhr-Universität Bochum for present support. I am also grateful to all the pioneers in carbenoid and s-block metal chemistry for their inspiring work and developments made over the years.Notes and references
- P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed.
- For reviews, see: (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (b) D. W. Stephan, Dalton Trans., 2009, 3129 RSC; (c) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535 RSC.
- For examples, see: (a) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS PubMed; (b) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS PubMed; (c) T. W. Hudnall, J. P. Moerdyk and C. W. Bielawski, Chem. Commun., 2010, 46, 4288 RSC; (d) F. Lavigne, E. Maerten, G. Alcaraz, V. Branchadell, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2012, 51, 2489 CrossRef CAS PubMed; (e) A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2012, 134, 1471 CrossRef CAS PubMed; (f) D. Schmidt, J. H. J. Berthel, S. Pietsch and U. Radius, Angew. Chem., Int. Ed., 2012, 51, 8881 CrossRef CAS PubMed; (g) G. D. Frey, J. D. Masuda, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 9444–9447 CrossRef CAS PubMed.
- H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323 CrossRef CAS.
- G. L. Closs and R. A. Moss, J. Am. Chem. Soc., 1962, 86, 4042 CrossRef.
- G. L. Closs and L. E. Closs, Angew. Chem., 1962, 74, 431 CrossRef CAS.
- (a) P. Fritsch, Liebigs Ann. Chem., 1894, 279, 319 CrossRef; (b) W. P. Buttenberg, Liebigs Ann. Chem., 1894, 279, 324 CrossRef; (c) H. Wiechell, Liebigs Ann. Chem., 1894, 279, 337 CrossRef.
- We'd like to distinguish here between main group metal and transition metal carbenoids. Historically, the definition of carbenoids included species of the general composition R2C(X)M with M being an s-block metal or zinc. In the last years, also transition metal compound which possess carbene-like reactivity have been described as carbenoids. These compounds will not be covered here. For recent reviews, see: (a) N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. D. Bruin, Catal. Sci. Technol., 2011, 1, 153 RSC; (b) E. Jellema, A. L. Jongerius, J. N. H. Reek and B. D. Bruin, Chem. Soc. Rev., 2010, 39, 1706 RSC; (c) H. M. L. Davies and S. J. Hedley, Chem. Soc. Rev., 2007, 36, 1109 RSC; (d) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC; (e) R. Beckhaus, Angew. Chem., 1997, 109, 694 CrossRef; (f) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (g) P. Müller, Acc. Chem. Res., 2004, 37, 243 CrossRef PubMed.
- G. Boche and J. C. W. Lohrenz, Chem. Rev., 2001, 101, 697 CrossRef CAS PubMed.
- Recent reviews on carbenoids: (a) V. Capriati and S. Florio, Chem. – Eur. J., 2010, 16, 4152 CrossRef CAS PubMed; (b) M. Pasco, N. Gilboa, T. Mejuch and I. Marek, Organometallics, 2013, 32, 942 CrossRef CAS; (c) T. Satoh, Chem. Soc. Rev., 2007, 36, 1561 RSC; (d) R. Knorr, Chem. Rev., 2004, 104, 3795–3849 CrossRef CAS PubMed; (e) V. Capriati, in Modern Lithium Carbenoid Chemistry, Contemporary Carbene Chemistry, ed. R. A. Moss and M. P. Doyle, Wiley, New York, 2014, ch. 11 Search PubMed; (f) S. Molitor and V. H. Gessner, Synlett, 2015, 861 CAS; (g) I. Marek, Tetrahedron, 2002, 58, 9463 CrossRef CAS.
- (a) G. Boche, M. Marsch, A. Müller and K. Harms, Angew. Chem., Int. Ed. Engl., 1993, 32, 1032 CrossRef; (b) G. Boche, K. Harms, M. Marsch and A. Müller, J. Chem. Soc., Chem. Commun., 1994, 1393 RSC.
- D. Seebach, R. Hässig and J. Gabriel, Helv. Chim. Acta, 1983, 66, 308 CrossRef CAS.
- G. Boche, F. Bosold, H. Hermann, M. Marsch, K. Harms and J. C. W. Lohrenz, Chem. – Eur. J., 1998, 4, 814 CrossRef CAS.
- For comparison, see: (a) G. Köbrich and H. R. Merkle, Chem. Ber., 1966, 99, 1782 CrossRef; (b) A. Müller, M. Marsch, K. Harms, J. C. W. Lohrenz and G. Boche, Angew. Chem., Int. Ed. Engl., 1996, 35, 1518–1520 CrossRef; (c) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 1529–1535 CrossRef CAS; (d) D. C. Kappeler and F. Hammerschmidt, J. Am. Chem. Soc., 2008, 130, 2329–2335 CrossRef PubMed.
- S. Molitor and V. H. Gessner, Angew. Chem., Int. Ed., 2016, 55, 7712 CrossRef CAS PubMed.
- (a) G. Köbrich and R. H. Fischer, Chem. Ber., 1968, 101, 3208–3218 CrossRef; (b) G. Köbrich and H. Papp, Z. Naturforsch., 1963, 186, 1125 Search PubMed; (c) G. Köbrich, H. Papp and I. Hornke, Tetrahedron Lett., 1964, 1131 Search PubMed; (d) G. Kobrich, H. Papp and I. Hornke, Chem. Ber., 1967, 100, 961 CrossRef.
- H. Hermann, J. C. W. Lohrenz, A. Kühn and G. Boche, Tetrahedron Lett., 2000, 56, 4109 CrossRef CAS.
- T. Clark and P. V. R. Schleyer, J. Chem. Soc., Chem. Commun., 1979, 88 Search PubMed.
- For lithiated amines, see: (a) V. H. Gessner and C. Strohmann, J. Am. Chem. Soc., 2008, 130, 14412 CrossRef CAS PubMed; (b) C. Strohmann and V. H. Gessner, Chem. – Asian J., 2008, 3, 1929 CrossRef CAS PubMed; (c) C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281 CrossRef CAS PubMed; (d) C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 4566 CrossRef CAS PubMed; (e) H. Ahlbrecht, J. Harbach, P. Hauck and H.-O. Kalinowski, Chem. Ber., 1992, 125, 1753 CrossRef CAS; (f) I. Kamps, D. Bojer, S. A. Hayes, R. J. F. Berger, B. Neumann and N. W. Mitzel, Chem. – Eur. J., 2009, 15, 11123 CrossRef CAS PubMed; (g) D. Bojer, I. Kamps, X. Tian, A. Hepp, T. Pape, R. Fröhlich and N. W. Mitzel, Angew. Chem., Int. Ed., 2007, 46, 4176 CrossRef CAS PubMed.
- (a) P. O'Brien, C. M. Rosser and D. Caine, Tetrahedron, 2003, 59, 9779 CrossRef; (b) P. Müller, D. Riegert and G. Bernardinelli, Helv. Chim. Acta, 2004, 87, 227 CrossRef; (c) J. Huang and P. O'Brien, Chem. Commun., 2005, 5696 RSC; (d) D. M. Hodgson, C. D. Bray and N. D. Kindon, Org. Lett., 2005, 7, 2305 CrossRef CAS PubMed; (e) D. M. Hodgson and S. M. Miles, Angew. Chem., Int. Ed., 2006, 45, 935 CrossRef CAS PubMed.
- (a) D. Seebach, Chem. Ber., 1972, 105, 487 CrossRef CAS; (b) M. Nitsche, D. Seebach and A. K. Beck, Chem. Ber., 1978, 111, 3644 CrossRef CAS.
- T. Rüffer, C. Bruhn, A. H. Maulitz, D. Ströhl and D. Steinborn, Organometallics, 2000, 19, 2829 CrossRef.
- T. Wagner, J. Lange, D. Grote, W. Sander, E. Schaumann, G. Adiwidjaja, A. Adam and J. Kopf, Eur. J. Org. Chem., 2009, 5198 CrossRef CAS.
- (a) G. Ludwig, D. Ströhl, H. Schmidt and D. Steinborn, Inorg. Chim. Acta, 2015, 429, 30 CrossRef CAS; (b) G. Ludwig, T. Rüffer, A. Hoppe, T. Walther, H. Lang, S. G. Ebbinghaus and D. Steinborn, Dalton Trans., 2015, 44, 5323 RSC.
- G. Köbrich and R. H. Fischer, Chem. Ber., 1968, 101, 3208 CrossRef.
- T. Cantat, X. Jacques, L. Ricard, X. F. Le Goff, N. Mézailles and P. Le Floch, Angew. Chem., Int. Ed., 2007, 46, 5947 CrossRef CAS PubMed.
- (a) C. Kupper, S. Molitor and V. H. Gessner, Organometallics, 2014, 33, 347 CrossRef CAS; (b) J. Becker and V. H. Gessner, Dalton Trans., 2014, 43, 4320 RSC; (c) K.-S. Feichtner and V. H. Gessner, Dalton Trans., 2014, 43, 14399 RSC.
- (a) H. Heuclin, S. Y.-F. Ho, X. F. Le Goff, C.-W. So and N. Mézailles, J. Am. Chem. Soc., 2013, 135, 8774 CrossRef CAS PubMed; (b) S. Y.-F. Ho, C.-W. So, N. Saffon-Merceron and N. Mézailles, Chem. Commun., 2015, 51, 2107 RSC.
- J. Konu and T. Chivers, Chem. Commun., 2008, 4995 RSC.
- For a review on alkenyllithium compounds: M. Braun, Angew. Chem., Int. Ed., 1998, 37, 430 CrossRef.
- (a) E. Niecke, P. Becker, M. Nieger, D. Stalke and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 1849 CrossRef CAS; (b) E. Niecke, M. Nieger, O. Schmidt, D. Gudat and W. W. Schoeller, J. Am. Chem. Soc., 1999, 121, 519 CrossRef CAS.
- (a) G. Köbrich, H. R. Merkle and H. Trapp, Tetrahedron Lett., 1965, 15, 969 CrossRef; (b) G. Köbrich and H. Trapp, Chem. Ber., 1966, 99, 670 CrossRef; (c) G. Köbrich and H. Trapp, Z. Naturforsch., B: J. Chem. Sci., 1963, 18, 1125 Search PubMed.
- F. M. Perna, A. Salomone, M. Dammacco, S. Florio and V. Capriati, Chem. – Eur. J., 2011, 17, 8216 CrossRef CAS PubMed.
- R. M. Magid and J. G. Welch, Tetrahedron Lett., 1967, 17, 2619 CrossRef.
- R. Tarhouni, B. Kirschleger, M. Rambaud and J. Villieras, Tetrahedron Lett., 1984, 25, 835–838 CrossRef CAS.
- (a) K. M. Sadhu and D. S. Matteson, Tetrahedron Lett., 1986, 27, 795 CrossRef CAS; (b) T. J. Michnick and D. S. Matteson, Synlett, 1991, 631 CrossRef CAS.
- (a) V. Pace, L. Castoldi and W. Holzer, J. Org. Chem., 2013, 78, 7764 CrossRef CAS PubMed; (b) V. Pace, W. Holzer, G. Verniest, A. R. Alcántara and N. De Kimpe, Adv. Synth. Catal., 2013, 355, 919 CrossRef CAS.
- D. Seyferth, R. L. Lambert Jr. and E. M. Hanson, J. Organomet. Chem., 1970, 24, 647 CrossRef CAS.
- G. Cainelli, N. Tangari and A. U. Ronchi, Tetrahedron, 1972, 28, 3009 CrossRef CAS.
- For examples, see (a) V. Pace, L. Castoldi and W. Holzer, Chem. Commun., 2013, 49, 8383 RSC; (b) V. Pace, L. Castoldi and W. Holzer, Adv. Synth. Catal., 2014, 356, 1761 CrossRef CAS; (c) K. M. Sadhu and D. S. Matteson, Organometallics, 1985, 4, 1687 CrossRef CAS; (d) P. R. Blakemore, S. P. Marsden and H. D. Vater, Org. Lett., 2006, 8, 773 CrossRef CAS PubMed.
- S. Dixon, S. M. Fillery, A. Kasatkin, D. Norton, E. Thomas and R. J. Whitby, Tetrahedron, 2004, 60, 1401 CrossRef CAS.
- (a) T. Satoh and K. Takano, Tetrahedron, 1996, 52, 2349 CrossRef CAS; (b) T. Satoh, K. Takano, H. Ota, H. Someya, K. Matsuda and M. Koyama, Tetrahedron, 1998, 54, 5557 CrossRef CAS . See also ref. 4c.
- For examples, see: (a) A. L. Barsamian and P. R. Blakemore, Organometallics, 2011, 31, 19 CrossRef; (b) C. R. Emerson, L. N. Zakharov and P. R. Blakemore, Chem. – Eur. J., 2013, 19, 16342 CrossRef CAS PubMed; (c) X. Sun and P. R. Blakemore, Org. Lett., 2013, 15, 4550 Search PubMed.
- P. Schröter and V. H. Gessner, Chem. – Eur. J., 2012, 18, 11223 CrossRef PubMed.
- (a) D. Seebach, H. Siegel, K. Müllen and K. Hiltbrunner, Angew. Chem., 1979, 91, 844 CrossRef CAS; (b) H. Siegel, K. Hiltbrunner and D. Seebach, Angew. Chem., 1979, 91, 845 CrossRef CAS; (c) D. Seebach, H. Siegel, J. Gabriel and R. Hässig, Helv. Chim. Acta, 1980, 63, 2046 CrossRef CAS; (d) D. Seebach, R. Hässig and J. Gabriel, Helv. Chim. Acta, 1983, 66, 308 CrossRef CAS; (e) D. Seebach, J. Gabriel and R. Hässig, Helv. Chim. Acta, 1984, 67, 1083 CrossRef CAS.
- H. Günther, in Advanced Applications of NMR to Organometallic Chemistry, ed. M. Gielen, R. Willem and B. Wrackmeyer, Wiley, Chichester, 1996, pp. 247–290 Search PubMed.
- (a) D. Tapu, D. A. Dixon and C. Roe, Chem. Rev., 2009, 109, 3385 CrossRef CAS PubMed; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940 CrossRef PubMed.
- D. Seebach, R. Hässig and J. Gabriel, Helv. Chim. Acta, 1983, 66, 308 CrossRef CAS.
- A. Salomone, F. M. Perna, A. Falcicchio, S. O. N. Lill, A. Moliterni, R. Michel, S. Florio, D. Stalke and V. Capriati, Chem. Sci., 2014, 5, 528 RSC.
- V. Schulze, R. Löwe, S. Fau and R. W. Hoffmann, J. Chem. Soc., Perkin Trans. 2, 1998, 464 Search PubMed.
- S. E. Denmark, J. P. Edwards and S. R. Wilson, J. Am. Chem. Soc., 1991, 113, 723 CrossRef CAS.
- A. B. Charette, J.-F. Marcoux, C. Molinaro, A. Beauchemin, C. Brochu and É. Isabel, J. Am. Chem. Soc., 2000, 122, 4508 CrossRef CAS.
- A. Voituriez, L. E. Zimmer and A. B. Charette, J. Org. Chem., 2010, 75, 1244 CrossRef CAS PubMed.
- A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12160 CrossRef CAS PubMed.
- G. Boche, F. Bosold, J. C. W. Lohrenz, A. Opel and P. Zulauf, Chem. Ber., 1993, 126, 1873 CrossRef CAS.
- A more detailed summary on computational studies is given in ref. 3.
- (a) T. Clark and P. V. R. Schleyer, J. Chem. Soc., Chem. Commun., 1979, 883 RSC; (b) T. Clark, P. V. R. Schleyer, K. N. Houk and N. G. Rondan, J. Chem. Soc., Chem. Commun., 1981, 579 RSC; (c) B. T. Luke, J. A. Pople, P. V. R. Schleyer and T. Clark, Chem. Phys. Lett., 1983, 102, 148 CrossRef CAS; (d) P. V. R. Schleyer, T. Clark, A. J. Kos, G. W. Spitznagel, C. Rohde, A. Arad, K. N. Houk and N. G. Rondan, J. Am. Chem. Soc., 1984, 106, 6467 CrossRef CAS.
- R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2010, 39, 9091 RSC.
- (a) C. Rohde, T. Clark, E. Kaufmann and P. V. R. Schleyer, J. Chem. Soc., Chem. Commun., 1982, 882 RSC; (b) L. M. Pratt, B. Ramachandran, J. D. Xidos, C. J. Cramer and D. G. Truhlar, J. Org. Chem., 2002, 67, 7607 CrossRef CAS PubMed.
- L. M. Pratt, N. V. Nguỹên and L. T. Lê, J. Org. Chem., 2005, 70, 2294 CrossRef CAS PubMed.
- L. M. Pratt, S. Merry, S. C. Nguyen, P. Quanb and B. T. Thanh, Tetrahedron, 2006, 62, 10821 CrossRef CAS.
- (a) T. Kimura and T. Satoh, J. Organomet. Chem., 2012, 715, 1 CrossRef CAS; (b) T. Kimura and T. Satoh, Tetrahedron, 2013, 69, 6371 CrossRef CAS.
- S. Harder, J. Boersma, L. Brandsma, J. A. Kanters, W. Bauer, R. Pi, P. V. R. Schleyer, H. Schöllhorn and U. Thewalt, Organometallics, 1989, 8, 1688 CrossRef CAS.
- G. Boche, F. Bosold, P. Zulauf, M. Marsch, K. Harms and J. Lohrenz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1455 CrossRef.
- K. Sorger, W. Bauer, P. V. R. Schleyer and D. Stalke, Angew. Chem., Int. Ed. Engl., 1995, 34, 1594 CrossRef CAS.
- G. Boche, A. Opel, M. Marsch, K. Harms, F. Haller, J. C. W. Lohrenz, C. Thümmler and W. Koch, Chem. Ber., 1992, 125, 2265 CrossRef CAS.
- For review, see: V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320 CrossRef CAS PubMed.
- T. Baumgartner, P. Moors, M. Nieger, H. Hupfer and E. Niecke, Organometallics, 2002, 21, 4919 CrossRef CAS.
- T. Baumgartner, D. Gudat, M. Nieger, E. Niecke and T. J. Schiffer, J. Am. Chem. Soc., 1999, 121, 5953 CrossRef CAS.
- B. Waerder, S. Steinhauer, B. Neumann, H.-G. Stammler, A. Mix, Y. V. Vishnevskiy, B. Hoge and N. W. Mitzel, Angew. Chem., 2014, 53, 11640 CrossRef CAS PubMed.
- (a) A. A. Kolomeitsev, A. A. Kadyrov, J. Szczepkowska-Sztolcman, H. Milewska, G. Bissky, J. A. Barten and G.-V. Röschenthaler, Tetrahedron Lett., 2003, 44, 8273 CrossRef CAS; (b) D. M. Roddick, Chem. Eng. News, 1997, 75, 6 CrossRef CAS.
- (a) D. V. Graham, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and C. Talmard, Chem. Commun., 2006, 417 RSC; (b) V. L. Blair, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2008, 5426 RSC.
- (a) A. B. Charette, A. Beauchemin and S. Francoeur, J. Am. Chem. Soc., 2001, 123, 8139 CrossRef CAS PubMed; (b) G. Boche, F. Bosold, H. Hermann, M. Marsch, K. Harms and J. C. W. Lohrenz, Chem. – Eur. J., 1998, 4, 814 CrossRef CAS; (c) F. A. Akkerman, R. Kickbusch and D. Lentz, Chem. – Asian J., 2008, 3, 719 CrossRef CAS PubMed; (d) I. Popov, S. Lindeman and O. Daugulis, J. Am. Chem. Soc., 2011, 133, 9286 CrossRef CAS PubMed.
- J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron, 1968, 24, 53 CrossRef CAS.
- (a) S. E. Denmark and J. P. Edwards, J. Org. Chem., 1991, 56, 6974 CrossRef CAS; (b) G. Wittig and F. Wingler, Chem. Ber., 1964, 97, 2146 CrossRef CAS.
- (a) S. E. Denmark, J. P. Edwards and S. R. Wilson, J. Am. Chem. Soc., 1992, 114, 2592 CrossRef CAS; (b) A. B. Charette, A. Beauchemin, S. Francoeur, F. Bélanger-Gariépy and G. D. Enright, Chem. Commun., 2002, 466 RSC; (c) A. B. Charette, C. Molinaro and C. Brochu, J. Am. Chem. Soc., 2001, 123, 12160 CrossRef CAS PubMed; (d) A. B. Charette, J.-F. Marcoux, C. Molinaro, A. Beauchemin, C. Brochu and E. Isabel, J. Am. Chem. Soc., 2000, 122, 4508 CrossRef CAS.
- S. E. Denmark, J. P. Edwards and S. R. Wilson, J. Am. Chem. Soc., 1991, 113, 723 CrossRef CAS.
- (a) A. Voituriez, L. E. Zimmer and A. B. Charette, J. Org. Chem., 2010, 75, 1244 CrossRef CAS PubMed; (b) M.-C. Lacasse, C. Poulard and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 12440 CrossRef CAS PubMed; (c) A. B. Charette, J.-F. Marcoux and F. Bélanger-Gariépy, J. Am. Chem. Soc., 1996, 118, 6792 CrossRef CAS.
- A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706 CrossRef CAS PubMed.
- (a) G. Köbrich and H. R. Merkle, Chem. Ber., 1967, 100, 3371 CrossRef; (b) G. Köbrich and H. R. Merkle, Angew. Chem., Int. Ed. Engl., 1967, 6, 74 CrossRef.
- (a) T. Hata, H. Kitagawa, H. Masai, T. Kurahashi, M. Shimizu and T. Hiyama, Angew. Chem., 2001, 113, 812 CrossRef; (b) T. Kurahashi, T. Hata, H. Masai, H. Kitagawa, M. Shimizu and T. Hiyama, Tetrahedron, 2002, 58, 6381 CrossRef CAS.
- For examples: (a) R. Rasappan and V. K. Aggarwal, Nat. Chem., 2014, 6, 810 CrossRef CAS PubMed; (b) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (c) S. Roesner, D. J. Blair and V. K. Aggarwal, Chem. Sci., 2015, 6, 3718 RSC; (d) Y. Fujioka and H. Amii, Org. Lett., 2008, 10, 769 CrossRef CAS PubMed; (e) P. R. Blakemore, S. P. Marsden and H. D. Vater, Org. Lett., 2006, 8, 773 CrossRef CAS PubMed; (f) P. R. Blakemore and M. S. Burge, J. Am. Chem. Soc., 2007, 129, 3068 CrossRef CAS PubMed; (g) C. G. Watson, A. Balanta, T. G. Elford, S. Essafi, J. N. Harvey and V. K. Aggarwal, J. Am. Chem. Soc., 2014, 136, 17370 CrossRef CAS PubMed.
- For review see: D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed.
- J. L. Stymiest, G. Dutheuil, A. Mahmood and V. K. Aggarwal, Angew. Chem., Int. Ed., 2007, 46, 7491 CrossRef CAS PubMed.
- (a) C. A. Brown and V. K. Aggarwal, Chem. – Eur. J., 2015, 21, 13900 CrossRef CAS PubMed; (b) G. Dutheuil, M. P. Webster, P. A. Worthington and V. K. Aggarwal, Angew. Chem., Int. Ed., 2009, 48, 6317 CrossRef CAS PubMed; (c) M. Binanzer, G. Y. Fang and V. K. Aggarwal, Angew. Chem., Int. Ed., 2010, 49, 4264 CrossRef CAS PubMed.
- S. Molitor and V. H. Gessner, Chem. – Eur. J., 2013, 19, 11858 CrossRef CAS PubMed.
- G. Märkl and A. Merz, Tetrahedron Lett., 1971, 1269 CrossRef.
- S. Molitor, J. Becker and V. H. Gessner, J. Am. Chem. Soc., 2014, 136, 15517 CrossRef CAS PubMed.
- V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC.
- (a) E. Negishi, K. Akiyoshi, B. O'Connor, K. Takagi and G. Wu, J. Am. Chem. Soc., 1989, 111, 3089 CrossRef CAS; (b) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 1999, 40, 9353 CrossRef CAS; (c) E. Thomas, A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 2006, 47, 9181 CrossRef CAS.
- E. Thomas, S. Dixon and R. J. Whitby, Tetrahedron, 2007, 63, 11686 CrossRef CAS.
- I. R. Baldwin and R. J. Whitby, Chem. Commun., 2003, 2786 RSC.
- F. J. Feher, D. D. Gergens and J. W. Ziller, Organometallics, 1993, 12, 2810 CrossRef CAS.
- E. Poverenov and D. Milstein, Chem. Commun., 2007, 3189 RSC.
- (a) S. Molitor, K.-S. Feichtner, C. Kupper and V. H. Gessner, Chem. – Eur. J., 2014, 20, 10752 CrossRef CAS PubMed; (b) V. H. Gessner, Organometallics, 2011, 30, 4228 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |