
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA six-step total synthesis of α-thujone and d6-α-thujone, enabling facile access to isotopically labelled metabolites†
Irene
Thamm
a,
Johannes
Richers
b,
Michael
Rychlik
a and
Konrad
Tiefenbacher
*cd
aAnalytical Food Chemistry, Technische Universität München, Alte Akademie 10, 85354 Freising, Germany
bDepartment of Chemistry, Technische Universität München, Lichtenbergstraße 4, 85747 Garching, Germany
cDepartment of Chemistry, University of Basel, St. Johannsring 19, CH-4056 Basel, Switzerland. E-mail: konrad.tiefenbacher@unibas.ch
dDepartment of Biosystems Science and Engineering, ETH Zürich, Mattenstrasse 26, CH-4058 Basel, Switzerland. E-mail: tkonrad@ethz.ch
First published on 12th September 2016
Abstract
The short synthesis of α-thujone relies on the functionalization of the readily available dimethylfulvene. Furthermore, the three main metabolites of the natural product were also synthesized. Since d6-acetone can be used as a starting material, the route developed allows for the facile incorporation of isotopic labels which are required for detecting and quantifying trace amounts via GC/MS analysis.
A great variety of plants produce the monoterpenes α-thujone (1a) and β-thujone (2, Fig. 1), which therefore are present in diverse herbal products.1 The most famous product containing thujone is certainly absinth, produced from wormwood. It had been a popular spirit drink in the 19th century but later was prohibited due to concerns about its toxicity.2 It was connected to severe health problems, including hallucinations, depressions, convulsions, blindness and mental deterioration. More recent studies propose that most of these effects were caused by alcohol intoxication.2 Nevertheless, thujone is neurotoxic and was shown to inhibit the gamma-aminobutyric acid A (GABAA) receptor, which leads to excitations and convulsions at higher concentrations in animal studies.3 α-Thujone (1a) was shown to be more active than its β-isomer 2. The metabolism of thujone was investigated both in vitro and in vivo. 7-OH-α-thujone (3a) is clearly the major metabolite in in vitro studies.3In vivo studies, however, point to 2-OH-α-thujone (4a) and 4-OH-α-thujone (5a) as the main metabolites.4
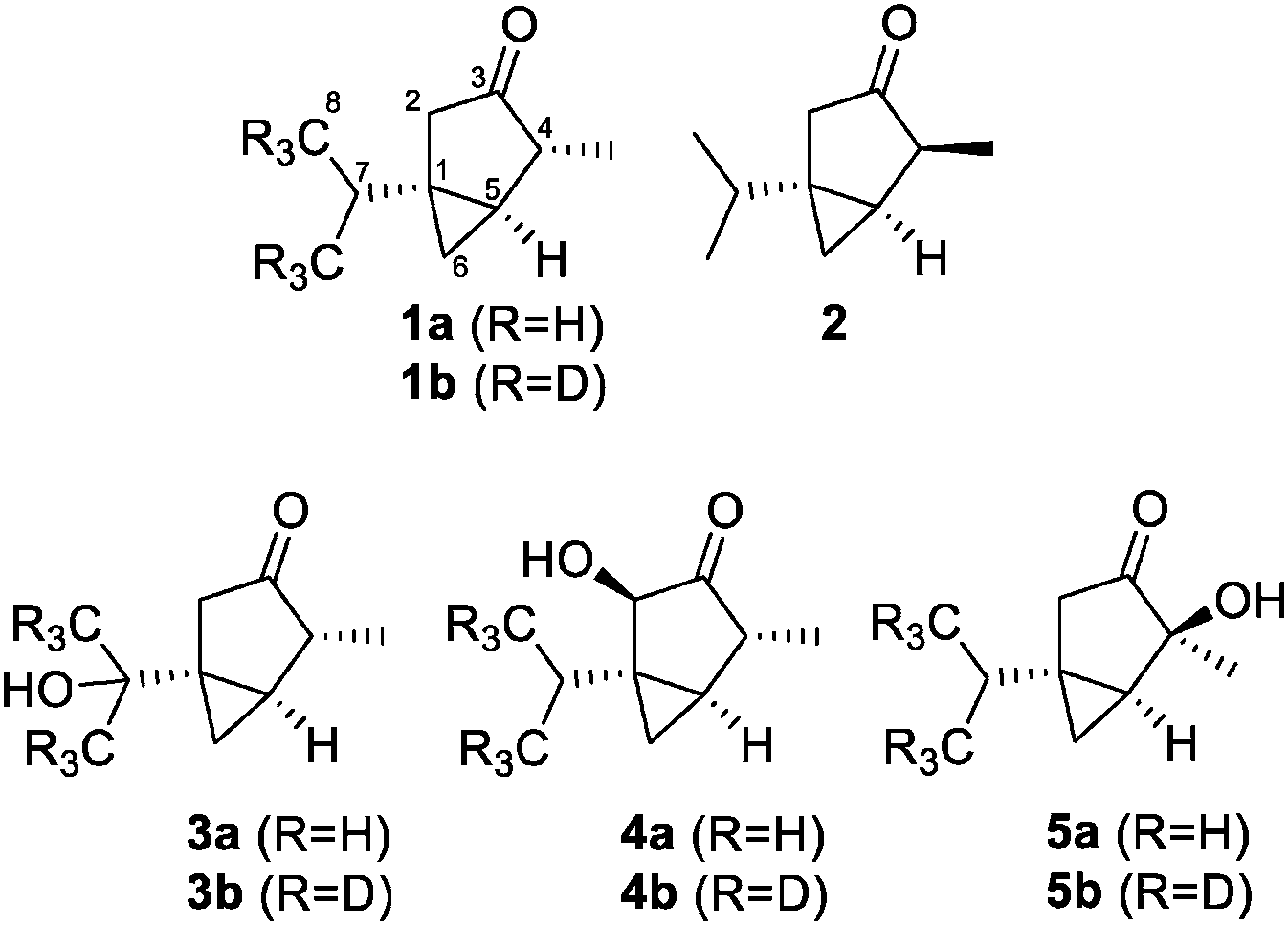 | ||
| Fig. 1 Structures of α-thujone (1a), β-thujone (2) and the major metabolites of α-thujone: 7-OH-α-thujone (3a), 2-OH-α-thujone (4a), and 4-OH-α-thujone (5a). | ||
The manufacture of thujone containing products is permitted again in the European Union but maximum limits have been imposed.5 To ensure accurate quantitation, to assess whether these products meet the requirements and additionally to better detect trace amounts of α-thujone and its major metabolites, access to isotopically labelled derivatives is required. Although the structure of the bicyclic monoterpene was elucidated in 1900 by Semmler,6 only one total synthesis has been reported so far.7 Oppolzer et al. prepared enantioenriched α-thujone over twelve steps from commercially available materials, utilizing an elegant palladium-catalyzed cyclization strategy. The route, however, does not allow for a facile introduction of inexpensive isotopic labels. Therefore, we developed and herein describe a novel six-step access to α-thujone, which enables the introduction of isotopic labels from inexpensive d6-acetone. The synthesized d6-thujone 1b thereafter is also functionalized to the most important metabolites 3b, 4b and 5b.
Our synthetic strategy is based on a late-stage oxidation of alcohol 6a/6b (Scheme 1), followed by a regio- and diastereoselective methylation at position C4. Such an approach seemed attractive since alcohol 6a/6b should be directly accessible from cyclopentenol 7a/7bvia Simmons–Smith cyclopropanation.8 Cyclopentenol 7a/7b can be traced back to the known dimethylfulvenes 8a and 8b, which are synthesized from cyclopentadiene and acetone.9 Therefore, the inexpensive d6-acetone can function as the source of the isotopic labels.
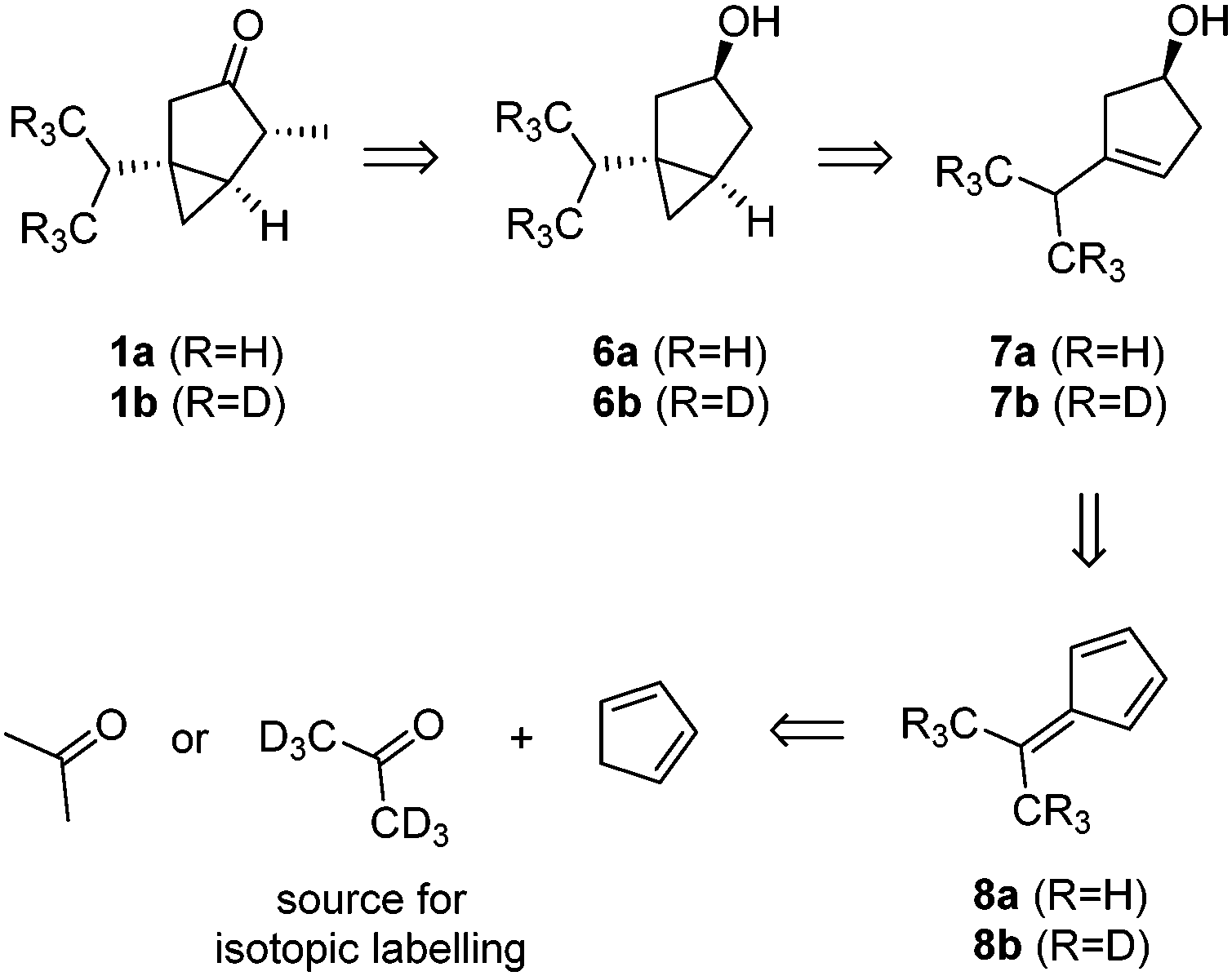 | ||
| Scheme 1 Retrosynthetic analysis of α-thujone (1a) and its deuterated derivative 1b. | ||
Our synthesis commenced with the formation of dimethylfulvene (Scheme 2). The preparation of 8a followed a procedure described by Little et al., utilizing pyrrolidine as a base.9a This procedure was not suitable for the preparation of the d6-dimethylfulvene 8b, since the deuterium label was partially removed, presumably via enamine formation. Therefore, the procedure introduced by the group of Gajewski was utilized to prepare d6-dimethylfulvene 8b.9b Using n-butyllithium as a base, a deuteration degree of 95.5% was achieved. The dimethylfulvenes formed were reduced with lithium aluminium hydride to yield a mixture of cyclopentadienes. It is known that related mixtures can be converted convergently to a single alcohol product via the hydroboration/oxidation sequence.10 Indeed, such a procedure yielded, after purification by chromatography, alcohols 7a and 7b in 34% and 27% yield over three steps, respectively. Cyclopropanation was performed utilizing the Furukawa modification of the Simmons–Smith reaction.11 After oxidation utilizing 2-iodoxybenzoic acid (IBX) as an oxidant,12 ketones 10a/10b were obtained in 77% and 88% yield, respectively. The final alkylation step required careful optimization of the reaction conditions. It was found that multi-alkylated products could not be separated by flash chromatography. Therefore, the reaction conditions were optimized to produce 1a/1b selectively. The use of 1.0 eq. of potassium bis(trimethylsilyl)amide and methyl iodide, as well as the addition of N,N′-dimethylpropyleneurea, facilitated this task. Thujones 1a and 1b were obtained in 75% and 62% yield, respectively.
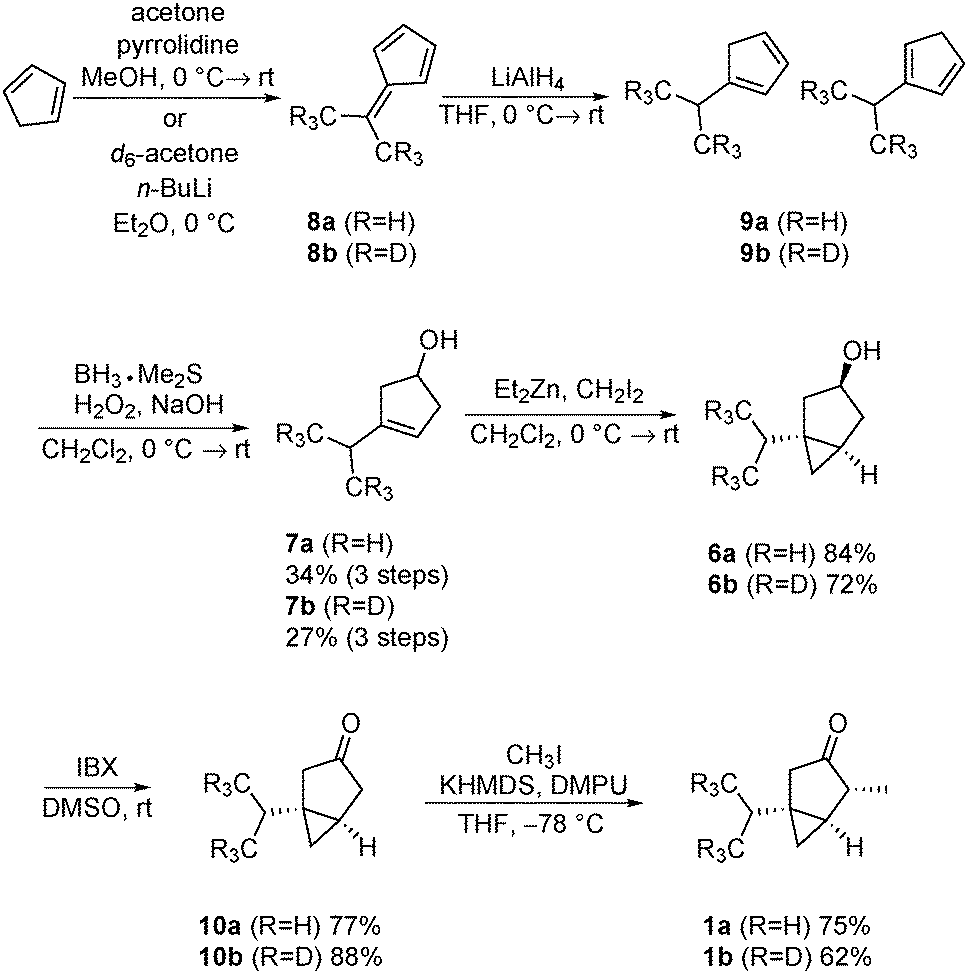 | ||
| Scheme 2 Synthesis of thujone 1a/1b. | ||
Although an optically active material is not required for use as an internal standard in GC/MS or LC/MS analysis, it may be helpful for other applications. Therefore, we investigated the possibility of performing the hydroboration of the cyclopentadienes 9a in an enantioselective fashion. Attempts with diisopinocampheylborane13 only lead to very low levels of enantiomeric enrichment (11% ee). It was therefore decided to turn to kinetic resolution. Pancreatin was utilized for the kinetic resolution of a structurally related cyclopentenol,14 and also proved to be effective in this case. Kinetic resolution provided acetate (+)-11a in high optical purity (95% ee) and acceptable conversion (35%) (Scheme 3). Alcohol (+)-7a, the enantiomer required, was not accessible efficiently via kinetic resolution. Time-consuming repetitions of kinetic resolutions were required to increase the conversions and enantiomeric excess. It turned out to be advantageous to work with acetate (+)-11 and invert its stereocenter via the Mitsunobu reaction. This three-step procedure delivered (+)-7a in high optical purity (95% ee). This material was converted to optically active thujone (−)-1a (95% ee) as described in Scheme 2 (see the ESI†).
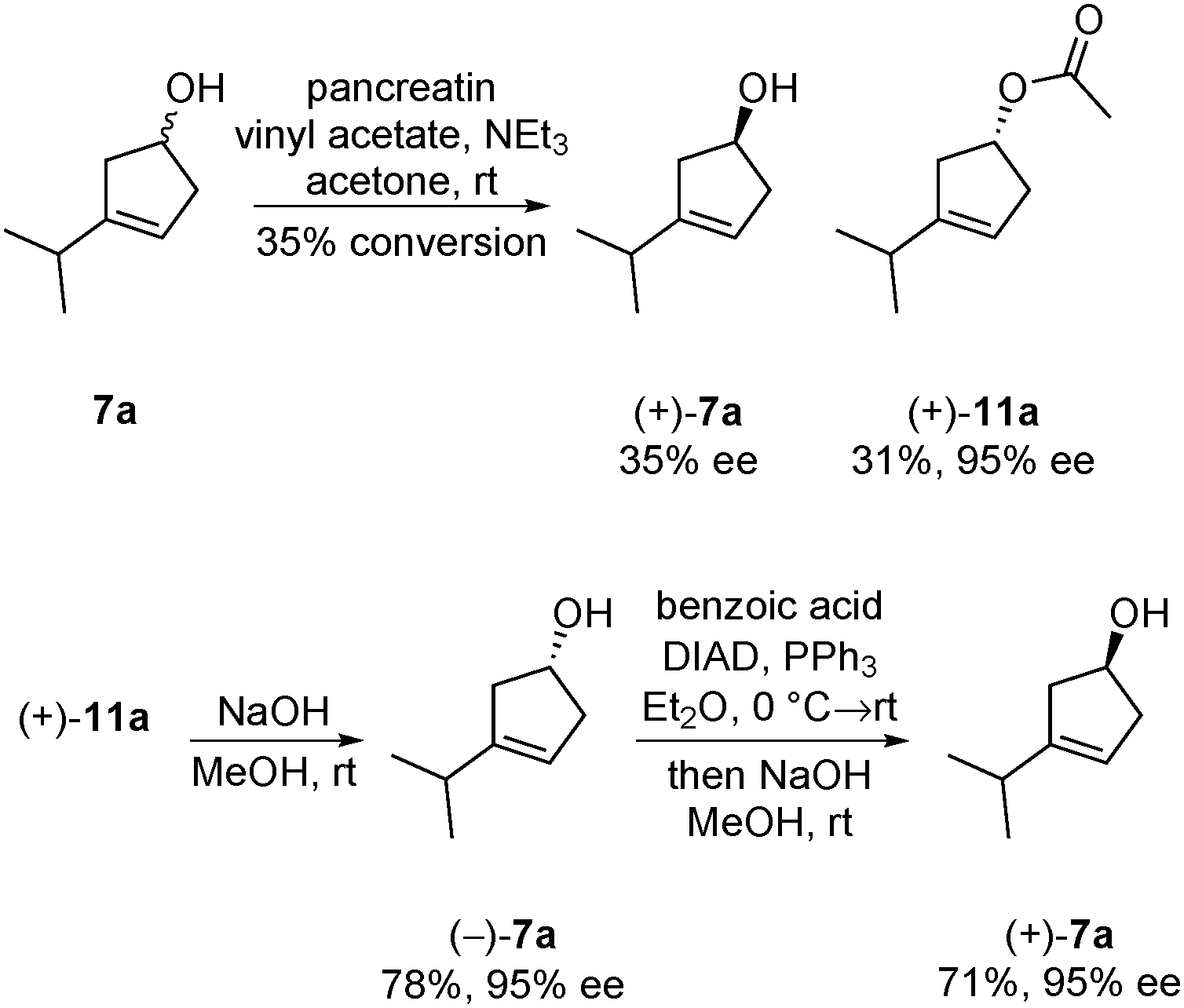 | ||
| Scheme 3 Kinetic resolution of (±)-3-isopropylcyclopent-3-en-1-ol and inversion of the chiral center via Mitsunobu reaction. | ||
After having developed a concise route to thujone (1a) and its isotopically labelled derivative 1b, we turned to the preparation of the most important metabolites. The oxidation of 1a to 7-OH-α-thujone (3a) was described in the literature utilizing ozone as the oxidant.15 However, these conditions lead to considerable overoxidation and a reduced yield of 3a (47%). After screening several oxidants, we found that methyl(trifluoromethyl)dioxirane16 (TFDO, 12) led to a clean conversion to 3a (Scheme 4). For the synthesis of the 4-hydroxy derivative 5a, we followed the procedure of the group of Casida.17 First, the enolacetate is formed by refluxing thujone in isopropenyl acetate under acidic conditions. In contrast to the literature, we observed the 2,3-enol acetate 13a as the main product (13a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14a = 8:2). Therefore, we decided to utilize this mixture to obtain both the 2-hydroxy and the 4-hydroxy derivative at once. The inseparable mixture was oxidized with 3-chloroperoxybenzoic acid. In both cases epoxidation was preferred from the convex bottom face, delivering after migration of the acetate group the desired diastereoisomers 15 and 16. The isomers were separated by chromatography, and subsequently deprotected to yield the metabolites 4 and 5, respectively. Deprotection of 15 was successful only under acidic conditions, as regular basic hydrolysis led to decomposition of the material.
:14a = 8:2). Therefore, we decided to utilize this mixture to obtain both the 2-hydroxy and the 4-hydroxy derivative at once. The inseparable mixture was oxidized with 3-chloroperoxybenzoic acid. In both cases epoxidation was preferred from the convex bottom face, delivering after migration of the acetate group the desired diastereoisomers 15 and 16. The isomers were separated by chromatography, and subsequently deprotected to yield the metabolites 4 and 5, respectively. Deprotection of 15 was successful only under acidic conditions, as regular basic hydrolysis led to decomposition of the material.
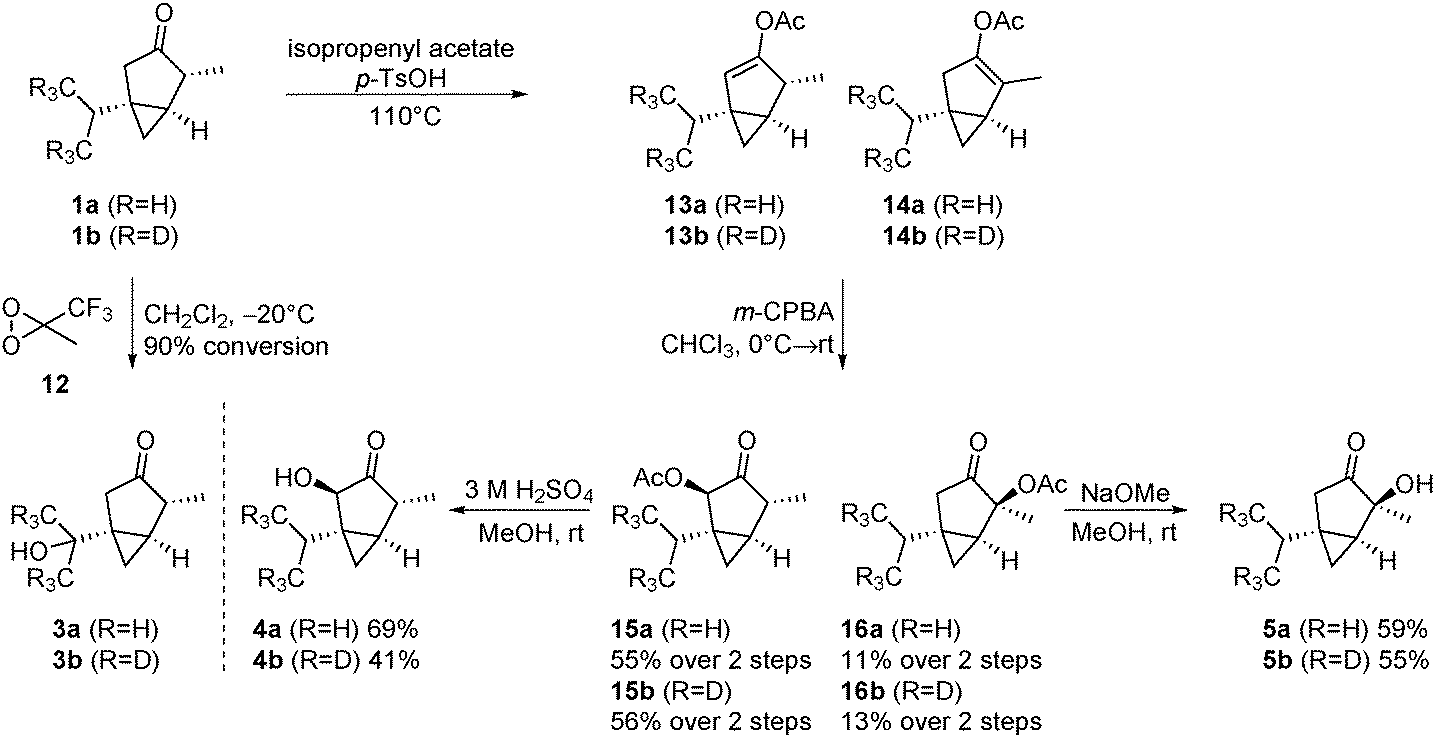 | ||
| Scheme 4 Synthesis of the hydroxylated main metabolites. | ||
In summary, we have developed a concise route to α-thujone, which relies on the functionalization of dimethylfulvene. The synthesis allows for the facile incorporation of inexpensive isotopic labels by utilizing d6-acetone as a starting material. Furthermore, the three main metabolites of α-thujone were prepared.
Notes and references
- O. Pelkonen, K. Abass and J. Wiesner, Regul. Toxicol. Pharmacol., 2013, 65, 100 CrossRef CAS PubMed.
- D. W. Lachenmeier, J. Emmert, T. Kuballa and G. Sartor, Forensic Sci. Int., 2006, 158, 1 CrossRef CAS PubMed.
- K. M. Hold, N. S. Sirisoma, T. Ikeda, T. Narahashi and J. E. Casida, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3826 CrossRef CAS PubMed.
- K. M. Höld, N. S. Sirisoma and J. E. Casida, Chem. Res. Toxicol., 2001, 14, 589–595 CrossRef.
- European Council Directive No. 88/388/EEC, 22 June 1988 http://ec.europa.eu/food/fs/sfp/addit_flavor/flav09_en.pdf (accessed on 23.11.2015).
- F. W. Semmler, Ber. Dtsch. Chem. Ges., 1900, 33, 275–277 CrossRef CAS.
- W. Oppolzer, A. Pimm, B. Stammen and W. E. Hume, Helv. Chim. Acta, 1997, 80, 623 CrossRef CAS.
- (a) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323 CrossRef CAS; (b) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- (a) K. J. Stone and R. D. Little, J. Org. Chem., 1984, 49, 1849 CrossRef CAS; (b) J. J. Gajewski, G. C. Paul, M. J. Chang and A. M. Gortva, J. Am. Chem. Soc., 1994, 116, 5150 CrossRef CAS.
- S. Collins, Y. Hong, M. Kataoka and L. Nguyen The, J. Org. Chem., 1990, 55, 3395 CrossRef CAS.
- J. Furukawa, N. Kawabata and J. Nishimura, Tetrahedron Lett., 1966, 7, 3353 CrossRef.
- M. Frigerio and M. Santagostino, Tetrahedron Lett., 1994, 35, 8019 CrossRef CAS.
- H. C. Brown and B. Singaram, J. Org. Chem., 1984, 49, 945 CrossRef CAS.
- (a) M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen and C. Meier, Chem. – Eur. J., 2012, 18, 11046 CrossRef CAS PubMed; (b) O. R. Ludek and C. Meier, Synthesis, 2003, 2101 CAS.
- J. P. Kutney, K. Piotrowska, Y.-H. Chen, K.-P. N. Cheng, Z. Gao and S. J. Rettig, Can. J. Chem., 1990, 68, 1698 CrossRef CAS.
- R. Mello, M. Fiorentino, C. Fusco and R. Curci, J. Am. Chem. Soc., 1989, 111, 6749 CrossRef CAS.
- N. S. Sirisoma, K. M. Höld and J. E. Casida, J. Agric. Food Chem., 2001, 49, 1915 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc05376a |
| This journal is © The Royal Society of Chemistry 2016 |