
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Peripheral ligands as electron storage reservoirs and their role in enhancement of photocatalytic hydrogen generation†
Qing
Pan
a,
Leon
Freitag
b,
Tanja
Kowacs
c,
Jane C.
Falgenhauer
d,
Jeroen P.
Korterik
a,
Derck
Schlettwein
d,
Wesley R.
Browne
e,
Mary T.
Pryce
f,
Sven
Rau
c,
Leticia
González
*b,
Johannes G.
Vos
*f and
Annemarie
Huijser
*a
aMESA + Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: j.m.huijser@utwente.nl
bInstitute of Theoretical Chemistry, University of Vienna, Währinger Str. 17, 1090 Vienna, Austria. E-mail: leticia.gonzalez@univie.ac.at
cInstitute of Inorganic Chemistry Materials and Catalysis, University of Ulm, Albert-Einstein-Allee 11, 89081 Ulm, Germany
dInstitute of Applied Physics, Justus-Liebig-University, Ludwigstraße 23, 35390 Gießen, Germany
eStratingh Institute for Chemistry, University of Groningen, 9747 AG, Groningen, The Netherlands
fSRC for Solar Energy Conversion, School of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: han.vos@dcu.ie
First published on 27th June 2016
Abstract
The contrasting early-time photodynamics of two related Ru/Pt photocatalysts with very different photocatalytic H2 generation capabilities are reported. Ultrafast equilibration (535 ± 17 fs) creates an electron reservoir on the peripheral ligands of the ester substituted complex, allowing a dramatic increase in photocatalytic performance. This insight opens the way towards a novel design strategy for H2 generating molecular photocatalysts.
Hydrogen generating molecular photocatalysts that mimic natural photosynthesis have been studied intensively in recent years.1 Since the first reports on H2 evolving Ru/M (M = Pt or Pd) bimetallic photocatalysts independently published by Sakai and Rau in 2006,2 many efforts have been made to increase the H2 generation efficiency in terms of both the turn-over number (TON) and turn-over frequency (TOF).3 Such optimisation studies are, however, complex since the photocatalytic efficiency is the result of a sequence of photophysical and photochemical processes occurring in a broad window of time scales. A sacrificial agent such as triethylamine (TEA) is required to regenerate the photosensitiser by electron donation to the formally oxidised Ru(III) centre. This process however also results in the formation of radicals, which may lead to side reactions by reacting with solvents (Section S5, ESI†). The presence of TEA, thus, prevents photophysical studies under catalytic conditions. However, earlier studies showed that the early-time (fs–ns) photodynamics under non-catalytic conditions provide important mechanistic insight into the photocatalytic performance. For instance, it was concluded that the electron density distribution of the initial photoexcited state determines the H2 generation efficiency.4 Furthermore, the occurrence of intramolecular electron transfer in the ps time window was shown to be essential.5 Hence, understanding of the fundamental early-time photodynamics is an important first step towards the design of improved photocatalysts. Such knowledge also facilitates the investigation of subsequent processes occurring at later time scales. This communication will therefore focus on the early-time photodynamics.
The early-time photodynamics and photocatalytic efficiency have been shown to be dependent on the peripheral/bridging ligands and the catalytic centre.6 Resonance Raman studies on various Ru/Pt and Ru/Pd complexes indicate that photoexcitation leads to the population of singlet 1MLCT (metal-to-ligand charge transfer) states based on both the peripheral and bridging ligands.5a,b,6a The intertwined involvement of these components suggests the importance of dynamic communication between different functional groups especially in the excited states. For instance, inter-ligand electron transfer has been observed in RuPd (Fig. 1) on a ps time scale, competing with vibrational cooling of excited states localised on the peripheral ligands, limiting the charge transfer efficiency towards the bridging ligand.5a Functionalising the bridging ligand of a Ru/Pt photocatalyst with an electron withdrawing CN group was observed to accelerate electron transfer to the bridging ligand and the H2 generation rate.5c Based on these observations, it is commonly accepted that a favouring population of excited states localised on the bridging ligand is essential for efficient photocatalysis. Generally the bridging ligand is designed to facilitate electron transfer from the photosensitiser to the catalytic centre, and store the photogenerated electron prior to any subsequent photochemical reactions.4,6d However, a recent study shows that an additional charge localised on the bridging ligand of an electrochemically mono-reduced Ru/Pt catalyst prevents directional transfer of the second electron.7 Utilisation of triplet 3MLCT states localised on the peripheral ligands is envisioned to be a pathway to increasing the efficiency of the essential second electron transfer process.
 | ||
| Fig. 1 Structure of the photocatalysts RuPd, RuPt and EtOOCRuPt. The ester unsubstituted and substituted precursors are denoted as Ru and EtOOCRu, respectively. S = solvent (acetonitrile). See Section S3, ESI† for more details. | ||
Indeed, comparison of a series of Ru/Pt and Ru/Pd complexes raises several questions. Two closely related bimetallic assemblies based on the same di(pyridine-2-yl)pyrazine (2,5-dpp) bridging ligand are both inactive in photocatalytic hydrogen generation,3a,6b suggesting that 2,5-dpp is not a suitable bridging ligand. Remarkably, functionalisation with ester groups of the peripheral 2,2′-bipyridine (bpy) ligands leads to a complex showing a TON of 400.6b A similar effect was also observed for 2,3-dpp bridged complexes.6a,8 More recently, two related Ru/Pt complexes based on a 2,2′:5′,2′′-terpyridine (tpy) bridging ligand have been investigated by our groups (RuPt and EtOOCRuPt, Fig. 1). EtOOCRuPt shows a significantly improved performance (TON = 650 after 6 h of irradiation at 470 nm) as compared to RuPt (TON = 80) in the same reaction conditions.9 For RuPd, a TON of 120 was reported.6c Although hydrogen generation depends on other parameters as well, it becomes clear that the peripheral ligands play a central role. To the best of our knowledge, no photodynamical investigation has been reported to explain the beneficial effect of ester functionalised peripheral ligands of these complexes.
Intersystem crossing (ISC) to the triplet manifold in Ru(II)–polypyridyl complexes is known to occur within 100 fs after photoexcitation10 and hence it is the energetics of the triplet states that determine the early-time photodynamics. The geometries and spin density distribution at the minima of the lowest 3MLCT states (Fig. 2) calculated by density functional theory (DFT), indicate that the order of the 3MLCT states changes upon ester modification of the peripheral ligands (Table S3-2, ESI†). For Ru and RuPt, a tpy-based 3MLCT state is the lowest in energy, whereas for EtOOCRu and EtOOCRuPt an EtOOC-bpy-based state becomes the lowest. This trend is the same for the lowest unoccupied molecular orbitals calculated at the ground state geometry (Fig. S3-1, ESI†).
 | ||
| Fig. 2 Spin densities of the lowest triplet states of Ru, EtOOCRu, RuPt and EtOOCRuPt in acetonitrile. | ||
The time evolution of the population of the (EtOOC-)bpy and tpy-localised 3MLCT states of Ru and EtOOCRu has been studied by transient absorption (TA). Fig. 3 (and Fig. S4-1, ESI†) shows the TA spectra of Ru and EtOOCRu. The TA spectrum of Ru shows two bands around 370 nm and 420 nm, with the first due to excited state absorption (ESA) of the bpy radical anion and the second related to the tpy radical anion.5a,11 The ESA increase around 420 nm concomitant with a decrease at ca. 370 nm in time manifests internal conversion (IC) from a bpy-based to a tpy-based 3MLCT state occurring within ca. 20 ps. Vibrational cooling is likely competing with this process.5a
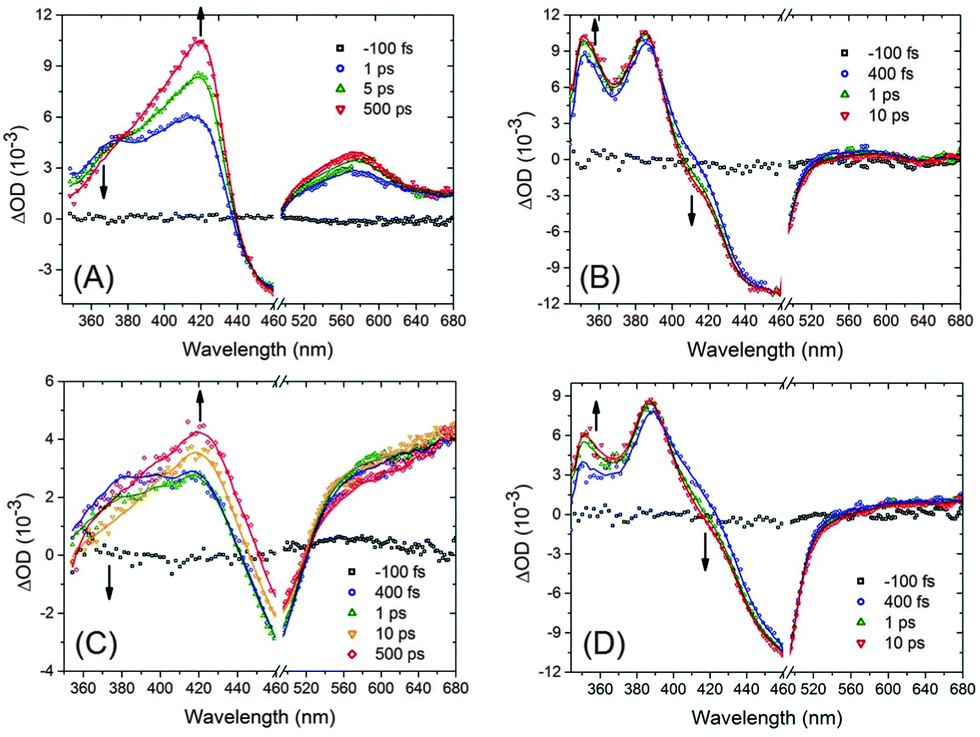 | ||
| Fig. 3 TA spectra of Ru (A), EtOOCRu (B), RuPt (C) and EtOOCRuPt (D) in anhydrous acetonitrile, including fits presented as solid curves.‡ | ||
The TA spectral features observed for EtOOCRu are different from those of Ru. An ESA band at ca. 350 nm increases in intensity within 1 ps, concomitant with a decrease of ESA at ca. 420 nm, as also evident from the kinetic traces provided in Fig. S4-1B (ESI†). Since the DFT calculations show that the 3MLCTEtOOC-bpy state is lower in energy than the 3MLCTtpy state, the observed dynamics likely reflect 3MLCTtpy → 3MLCTEtOOC-bpy IC. Based on this interpretation, the ESA bands at ca. 350 nm and 420 nm are assigned to the reduced EtOOC-bpy and tpy ligands, respectively. An additional ESA band is present at ca. 385 nm, which does not evolve in time significantly, likely due to the overlapping contributions from the nearby ESA bands around 350 nm and 420 nm. The observation of a similar band at ca. 385 nm for the homoleptic complex [Ru(EtOOC-bpy)3]2+ in acetonitrile12 indicates that this ESA band is likely associated to a reduced EtOOC-bpy ligand. The assignment of the ESA bands at both 350 nm and 385 nm to the EtOOC-bpy ligands is consistent with the species associated spectra (Fig. S4-3, ESI†) obtained from the photophysical model shown in Fig. S4-2 (ESI†). Further indirect support is provided by the differential absorption spectra under electrochemical reduction conditions (Fig. S2-3, ESI†). The above assignment suggests that, although ester functionalisation of the bpy ligands does not significantly alter the ESA position of the tpy ligand at ca. 420 nm, the bpy-based ESA band is split, possibly due to coupling between different ligands.§ The absence of spectral changes at times beyond a few ps (up to at least 6 ns, Fig. S4-4, ESI†) indicates that the formed 3MLCT manifold is long-lived and excited state equilibration between 3MLCTEtOOC-bpy and 3MLCTtpy states in EtOOCRu is ultrafast (Fig. S4-2, ESI†).
The TA spectral features of RuPt and EtOOCRuPt (Fig. 3) are similar to those of their mononuclear precursors. For RuPt, an ESA band at ca. 370 nm becomes less intense in time concomitant with a growth of an ESA at ca. 420 nm, indicating IC from bpy-localised 3MLCT states to tpy-based 3MLCT states. Again, as observed for the two monometallic precursors, the direction of IC observed for EtOOCRuPt is opposite to that of RuPt, indicating that cyclometalation by Pt does not change the direction of ultrafast intramolecular electron transfer.
A photophysical model of EtOOCRuPt established by target analysis of the TA data is shown in Fig. 4. The obtained species associated spectra are provided in Fig. S4-5 (ESI†). Photoexcitation leads to EtOOC-bpy and tpy-based singlet excited states, which undergo ultrafast ISC10 into EtOOC-bpy and tpy-Pt localised triplet states. The electron density at the bridge side moves on an ultrafast timescale to the EtOOC-bpy ligands (535 ± 17 fs). The backward electron transfer from EtOOC-bpy to tpy-Pt occurs at >3 ps. This equilibration results in only a minor population of the bridge based states.
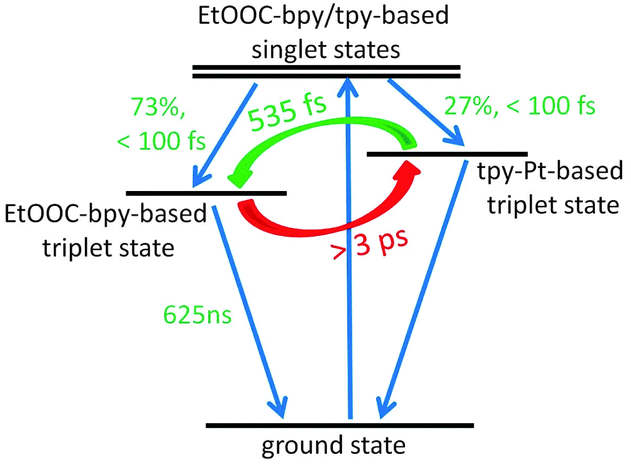 | ||
| Fig. 4 Simplified photophysical model of light-induced processes in EtOOCRuPt. | ||
The TA data discussed above allow interpretation of the time-resolved photoluminescence data obtained before.9 The excited state decay of Ru is mono-exponential (τ = 796 ± 6 ns), indicating that the various peripheral and bridging ligand based low lying 3MLCT states are thermally equilibrated.13 For EtOOCRu (τ = 1.05 ± 0.01 μs) and EtOOCRuPt (τ = 625 ± 31 ns), ultrafast excited state equilibration results in a major population of the 3MLCTEtOOC-bpy states, yielding a mono-exponential emission decay. However, for RuPt a bi-exponential decay (τ1 = 124 ± 58 ns, 15%; τ2 = 658 ± 19 ns, 85%) is observed. RuPd shows analogous features with the fast component more pronounced.5a The fast component observed for RuPt is possibly due to enhanced spin–orbit coupling caused by Pt increasing the radiative decay rate. Alternatively, it can be explained by an increased rigidity of the bridging ligand upon coordination with the Pt centre, possibly resulting in a lowering of the energy of the deactivating triplet metal-centred (3MC) level at the Ru moiety. The increased rigidity may also reduce the vibrational freedom of the ligand thereby increasing the difficulty of inter-ligand IC. This hypothesis is in agreement with DFT calculations showing that the root-mean-square deviation between the bpy and tpy structures increases upon bonding of the Pt centre (Table S3-2, ESI†). As a result, both 3MLCT states contribute to the photoluminescence, resulting in a bi-exponential decay.
As discussed above, an important effect of ester functionalisation of the peripheral bpy ligands involves a lowering in 3MLCT energy levels. A major consequence is an inversion in inter-ligand IC direction, causing an ultrafast (535 ± 17 fs) population of 3MLCT states localised on the peripheral ligands. This result seemingly indicates that electron density is moving in a disadvantageous direction. A priori, it could be anticipated that movement of electron density from the photosensitiser to the catalytic centre by population of the bridging ligand would be beneficial for photocatalytic H+ reduction. This strategy has been applied before by substitution of the bridge with an electron withdrawing CN group in a Ru/Pt photocatalyst (RuPt-CN), and improved electron transfer to the catalytic Pt moiety and hydrogen evolution were observed.5c However, for EtOOCRuPt the electron transfer direction is the opposite while at the same time a much higher efficiency is achieved for EtOOCRuPt compared to RuPt.9 Definitely, the present work provides the first photophysical evidence challenging the conventional design paradigm based on a bridging ligand acting as electron density storage reservoir.
Establishing a connection between storage of electron density at the peripheral ligands at early-times and the high H2 TON and TOF values is not straightforward considering the multiple photochemical reactions occurring at later times. However, it is important to consider two key parameters required for efficient H2 generation; a long excited state lifetime and efficient IC towards the bridging ligand. A long excited state lifetime is essential to allow multiple slow (diffusion controlled) processes to proceed. However, fast IC to the bridge does not guarantee a long lifetime. For instance, a negative side effect observed in RuPt-CN involved the recombination of 30–40% of the excited states within ∼1 ps.5c Deactivation induced by the cyclometalated Pt due to, e.g. strong spin–orbit coupling and additional nonradiative deactivation, is likely to reduce the bridge-based excited state lifetime.5a However, when the photoexcited electron is localised on the peripheral ligands, the relatively longer distance to the Pt centre makes the Pt induced deactivation channels less accessible. Furthermore, functionalisation of the peripheral ligands with ester groups stabilises the 3MLCT states, increasing the gap with the deactivating 3MC state.14 These effects result in a longer excited state lifetime. An analogous trend has been observed in Ru/Mn photocatalysts for oxygen evolution: the lifetime is reduced from 1400 ns to 23 ns upon Mn coupling for unsubstituted complexes, whereas for the ester substituted analogues the lifetimes remain almost independent of Mn coupling (>1000 ns).15 Most interestingly, when the ester group is also present at the bridge, the improvement achieved by ester functionalisation of the peripheral ligands is lost.16 Besides an improved excited state lifetime, populating the peripheral ligands by the first photogenerated electron may also have a significant impact on the potential energy surfaces relevant for the second photoelectron, promoting future investigation of this fundamental aspect.
In summary, the key observation in the present work is that ultrafast intramolecular equilibration resulting in storing the first photogenerated electron on the peripheral ligands at early times allows a considerable increase in photocatalytic efficiency. This observation cannot be explained within the traditional framework based on the bridging ligand acting as electron storage reservoir. An important advantage of 3MLCT states on the peripheral ligands relative to those on the bridge is their lower susceptibility to excited state deactivation induced by the catalytic centre. These results allow for the development of new design futures for the optimisation of molecular photocatalysts.
This work is supported by the Dutch Organisation for Scientific Research (NWO) and the EU-COST Action PERSPECT-H2O (STSM 16737). Glotaran17 was used for data analysis. Prof. Huib Bakker (AMOLF) is acknowledged for discussions.
Notes and references
- (a) L. C. Sun, L. Hammarström, B. Akermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36 RSC; (b) A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjo, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899 CrossRef CAS PubMed; (c) L. Hammarström, Acc. Chem. Res., 2015, 48, 840 CrossRef PubMed.
- (a) H. Ozawa, M. A. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926 CrossRef CAS PubMed; (b) S. Rau, B. Schafer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Gorls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215 CrossRef CAS PubMed.
- (a) H. Ozawa and K. Sakai, Chem. Commun., 2011, 47, 2227 RSC; (b) Y. Halpin, M. T. Pryce, S. Rau, D. Dini and J. G. Vos, Dalton Trans., 2013, 42, 16243 RSC.
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. Gonzalez, M. Schmitt, S. Rau and J. Popp, Angew. Chem., Int. Ed., 2010, 49, 3981 CrossRef CAS PubMed.
- (a) Q. Pan, F. Mecozzi, J. P. Korterik, D. Sharma, J. L. Herek, J. G. Vos, W. R. Browne and A. Huijser, J. Phys. Chem. C, 2014, 118, 20799 CrossRef CAS; (b) S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundström, M. Karnahl, M. Schwalbe, B. Schafer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15, 7678 CrossRef CAS PubMed; (c) C. V. Suneesh, B. Balan, H. Ozawa, Y. Nakamura, T. Katayama, M. Muramatsu, Y. Nagasawa, H. Miyasaka and K. Sakai, Phys. Chem. Chem. Phys., 2014, 16, 1607 RSC.
- (a) T. Kowacs, Q. Pan, P. Lang, L. O'Reilly, S. Rau, W. R. Browne, M. T. Pryce, A. Huijser and J. G. Vos, Faraday Discuss., 2015, 185, 143 RSC; (b) G. S. Bindra, M. Schulz, A. Paul, S. Soman, R. Groarke, J. Inglis, M. T. Pryce, W. R. Browne, S. Rau, B. J. Maclean and J. G. Vos, Dalton Trans., 2011, 40, 10812 RSC; (c) G. S. Bindra, M. Schulz, A. Paul, R. Groarke, S. Soman, J. L. Inglis, W. R. Browne, M. G. Pfeffer, S. Rau, B. J. MacLean, M. T. Pryce and J. G. Vos, Dalton Trans., 2012, 41, 13050 RSC; (d) M. Karnahl, C. Kuhnt, F. Ma, A. Yartsev, M. Schmitt, B. Dietzek, S. Rau and J. Popp, ChemPhysChem, 2011, 12, 2101 CrossRef CAS PubMed; (e) M. G. Pfeffer, B. Schafer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wachtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044 CrossRef CAS PubMed; (f) M. G. Pfeffer, T. Kowacs, M. Wachtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 6627 CrossRef CAS PubMed; (g) Q. Pan, F. Mecozzi, J. P. Korterik, J. G. Vos, W. R. Browne and A. Huijser, ChemPhysChem, 2016 DOI:10.1002/cphc.201600458.
- L. Zedler, J. Guthmuller, I. R. de Moraes, S. Kupfer, S. Krieck, M. Schmitt, J. Popp, S. Rau and B. Dietzek, Chem. Commun., 2014, 50, 5227 RSC.
- H. Ozawa, Y. Yokoyama, M. Haga and K. Sakai, Dalton Trans., 2007, 1197 RSC.
- T. Kowacs, L. O'Reilly, Q. Pan, A. Huijser, P. Lang, S. Rau, W. R. Browne, M. T. Pryce and J. G. Voss, Inorg. Chem., 2016, 55, 2685 CrossRef CAS PubMed.
- (a) N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275, 54 CrossRef CAS PubMed; (b) A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2006, 45, 3174 CrossRef CAS PubMed.
- (a) S. Wallin, J. Davidsson, J. Modin and L. Hammarstrom, J. Phys. Chem. A, 2005, 109, 4697 CrossRef CAS PubMed; (b) C. W. Stark, W. J. Schreier, J. Lucon, E. Edwards, T. Douglas and B. Kohler, J. Phys. Chem. A, 2015, 119, 4813 CrossRef CAS PubMed.
- B. H. Farnum, J. M. Gardner and G. J. Meyer, Inorg. Chem., 2010, 49, 10223 CrossRef CAS PubMed.
- D. Magde, M. D. Magde and E. C. Glazer, Coord. Chem. Rev., 2016, 306, 447 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759 CrossRef CAS.
- M. L. A. Abrahamsson, H. B. Baudin, A. Tran, C. Philouze, K. E. Berg, M. K. Raymond-Johansson, L. C. Sun, B. Akermark, S. Styring and L. Hammarström, Inorg. Chem., 2002, 41, 1534 CrossRef CAS PubMed.
- Y. H. Xu, G. Eilers, M. Borgstrom, J. X. Pan, M. Abrahamsson, A. Magnuson, R. Lomoth, J. Bergquist, T. Polivka, L. C. Sun, V. Sundström, S. Styring, L. Hammarström and B. Akermark, Chem. – Eur. J., 2005, 11, 7305 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1 Search PubMed.
- A. Staniszewski, W. B. Heuer and G. J. Meyer, Inorg. Chem., 2008, 47, 7062 CrossRef CAS PubMed.
- (a) P. Qu and G. J. Meyer, Langmuir, 2001, 17, 6720–6728 CrossRef CAS; (b) B. H. Farnum, J. J. Jou and G. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15628 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, additional spectroscopy and DFT results. See DOI: 10.1039/c6cc05222c |
| ‡ The fits for EtOOCRu and EtOOCRuPt are based on the photophysical models shown in Fig. S4-2 (ESI†) and Fig. 4. The fits for Ru and RuPt are obtained from global analysis using a sequential model.6g |
| § The ESA features of related heteroleptic complexes are known to depend on the combination of ligands. An ESA band at ca. 350 nm was found for [Ru(EtOOC-bpy)2BTL]2+ (BTL = 9′-[4,5-bis(cyanoethylthio)]-[1,3-dithiol-2-ylidene]-4′,5′-diazafluorene).18 For [Ru(EtOOC-bpy)2(2,3-dpp)PtI2]2+, an ESA band at ca. 385 nm was observed,6a while [Ru(EtOOC-bpy)(bpy)2]2+ manifests a broad ESA band below ca. 400 nm.19 |
| This journal is © The Royal Society of Chemistry 2016 |