
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blue-light activated rapid polymerization for defect-free bulk Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) crosslinked networks†
Abhishek U.
Shete
a,
Bassil M.
El-Zaatari
b,
Jonathan M.
French
a and
Christopher J.
Kloxin
*ab
aDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA. E-mail: cjk@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE 19716, USA
First published on 2nd August 2016
Abstract
A visible-light (470 nm wavelength) sensitive Type II photoinitiator system is developed for bulk Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reactions in crosslinked networks. The accelerated photopolymerization eliminates UV-mediated azide decomposition allowing for the formation of defect-free glassy networks which exhibit a narrow glass transition temperature.
The Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is one of the most widely implemented ‘click’ reactions.1 The wide range of conditions in which the bio-orthogonal CuAAC reaction can be applied makes it immensely useful for an array of chemical2 and material3,4 research application such as hydrogels,5–7 bioconjugation,8,9 surface modifications,10,11 and dendrimers.12,13 Typically, the reaction proceeds through an in situ generation of catalytic Cu(I) from Cu(II) using a reducing agent such as sodium ascorbate.14–16 In the last decade, several researchers have also generated Cu(I) using a photo-chemical route for a range of small molecule17,18 and polymerization19 applications. In the photo-CuAAC reaction, Cu(II) is reduced through a reaction with radical species produced via irradiation of either a Norrish Type I (α-cleavage) or Norrish Type II (electron transfer) photoinitiator.20–22 The generation of the Cu(I) catalyst using light enables temporal control; moreover, despite the potential of Cu(I) to diffuse through the system spatial control is also observed likely owing to Cu(I) interactions with the triazole product.19,23
Several favourable attributes of the photo-CuAAC reaction in neat (solventless) polymerizations of multifunctional azide and alkyne monomers are diminished by the formation of defects (bubbles). These defects formed upon polymerization under UV light compromise the structural integrity of the film. It is well recognized in the literature that the azide functional group exhibits susceptibility to photodecomposition, particularly under ultraviolet (UV) light exposure, forming a reactive nitrene species and emitting nitrogen gas.24–27 For example, aromatic azides are used in light-initiated azide–amine bioconjugation reactions, where azides photodecompose to form a reactive nitrene followed by ring expansion and reaction with the amine substrate.28,29 While the observation of bubble formation is notably absent in the literature, we herein report that bubbles do indeed form during the photopolymerization of a neat (solventless) crosslinked polymer. The formation and extent of bubbles depend on a number of experimental conditions, including the wavelength and intensity of light, sample thickness, and roughness of the sample preparing surface (presence of nucleation sites); however, the generation of nitrogen bubbles can be prevented entirely through the use of longer wavelength light in conjunction with an appropriately absorbance-matched photoinitiating system.
Inspired by the widely used blue-light activated methacrylate-based dental-restorative resins,30,31 we introduce an accelerated photo-CuAAC bulk polymerization system to obtain glassy crosslinked networks that are defect (bubble) free. The developed blue-light sensitive Type II photosensitizing system includes optimized concentrations of camphorquinone and a hydrogen-donating tertiary amine as an accelerator to afford polymer networks that are not only completely defect-free under all experimental conditions but also show rapid polymerization. This methodology to obtain bulk photo-CuAAC network additionally takes advantage of ‘dark polymerization’, wherein the network continues to crosslink after irradiation is ceased, owing to the persistence of catalytic Cu(I) species.32,33 The photo-CuAAC-based polymerization maintains a step-growth molecular weight evolution producing characteristic high functional group conversions with a narrow glass transition region.
Typically, bulk photopolymerized CuAAC network systems have been initiated with UV wavelength light (365 nm)34 or near-UV wavelengths (405 nm)35 using an appropriate absorbance-matched photoinitiator species. As discussed, continuous UV irradiation at these wavelengths can lead to the photodecomposition of azide functional groups present on the monomers. While azide photodecomposition is a side reaction compared to the azide–alkyne crosslinking reaction, defects (bubbles) are clearly observed in 0.5 mm thick films formed using monomers 1 and 2 (see Fig. 1A). Correspondingly, the CuAAC resins irradiated with near-UV (i.e., 405 nm) wavelength light contain qualitatively fewer bubbles but still compromise the integrity of the specimen.
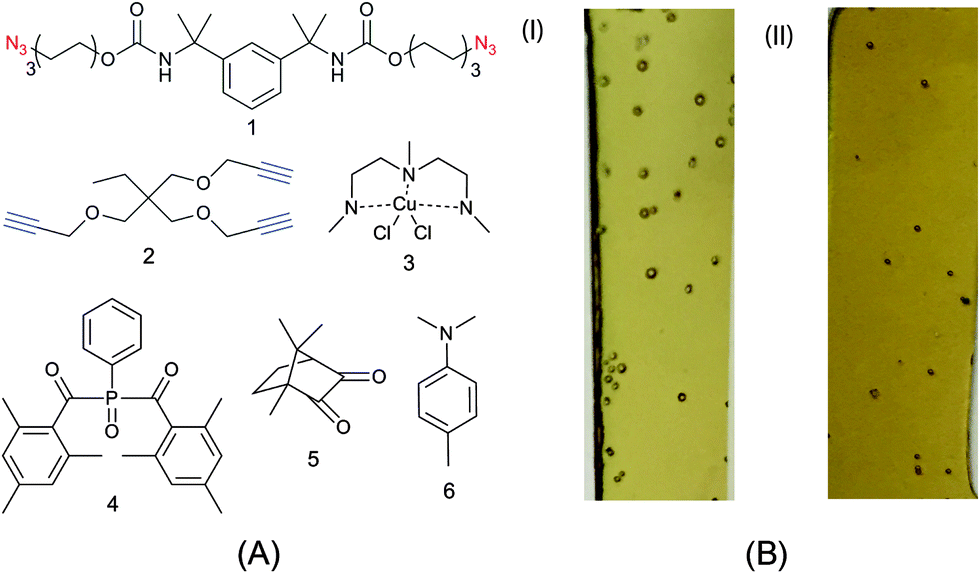 | ||
Fig. 1 (A) Monomers and reagents used in investigations. (B) Representative examples of 0.5 mm thick films formed using 365 nm (I) and 405 nm (II) wavelength light showing defect in the form of bubble specks. The specimens contained monomers bis(6-azidohexyl)(1,3-phenylenebis(propane-2,2-diyl))dicarbamate (1) and 1-(prop-2-ynyloxy)-2,2-bis(prop-2-ynyloxymethyl)butane (2) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of azide:alkyne functional group with 1.5 wt% Cu(II)-PMDETA complex (3) and 1 wt% Irgacure 819 (4) as the photoinitiator and were continuously irradiated for 10 minutes at an intensity of 40 mW cm−2. :1 ratio of azide:alkyne functional group with 1.5 wt% Cu(II)-PMDETA complex (3) and 1 wt% Irgacure 819 (4) as the photoinitiator and were continuously irradiated for 10 minutes at an intensity of 40 mW cm−2. | ||
For samples irradiated with 365 and 405 nm light, we have noted several experimental conditions that affect bubble formation in the photo-CuAAC network system shown in Fig. 1A. First, increasing light intensity and dose, which is necessary to have complete conversion in the depth of thick samples, increases the amount and size of bubbles in the system. Specifically, fewer bubbles were observed in thinner samples irradiated over a smaller timescale. Second, the surface roughness on which the CuAAC reaction is performed appears to affect bubble formation, presumably by providing additional nucleation sites. Nevertheless, the wavelength of light remains the main experimental condition that controls bubble formation (i.e., shorter wavelengths lead to increased bubble formation).
To support our hypothesis that azide photodecomposition is the source of the observed bubble defects, UV-Vis spectroscopy of the azide monomer (monomer 1) (1 mM in N,N-dimethylformamide; DMF) was performed under irradiation with UV-light (365 nm) and blue-light (470 nm) at an intensity of 40 mW cm−2. Fig. 2A shows the decrease in absorbance in the peak associated with azide at 290 nm for only monomer 1 in DMF irradiated using 365 nm UV light. The nitrogen gas produced was further verified using gas chromatography (GC) mass spectroscopy (see Fig. S1 in ESI†). In contrast, the azide absorbance remains unchanged under 470 nm blue-light confirming the stability of azide monomer under longer wavelength of similar intensity.
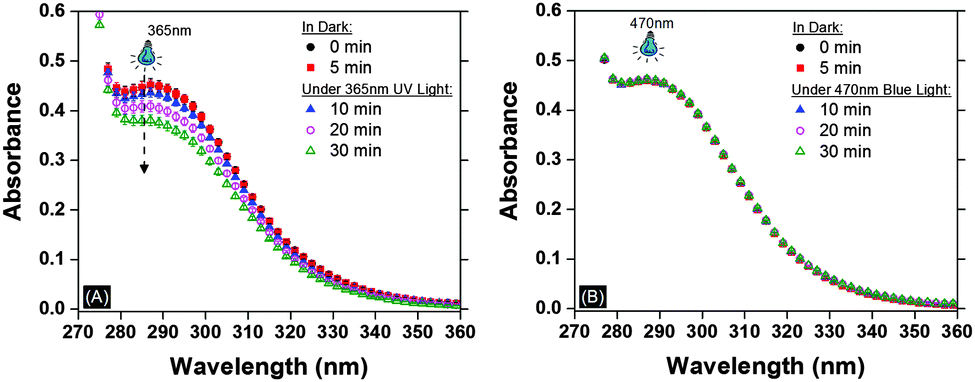 | ||
| Fig. 2 (A) The UV-Vis spectroscopy analysis of 1i.e. bis(6-azidohexyl)(1,3-phenylenebis(propane-diyl))dicarbamate in N,N-dimethylformamide (DMF). Absorbance spectrum of a 1 mM solution of monomer 1 in DMF in a cuvette of 10 mm thickness (↓) shows a decrease in the absorbance under 365 nm (UV) light of 40 mW cm−2 indicating an azide concentration decrease due to decomposition. (B) Absorbance is unchanged under 470 nm (blue) light of the same intensity showing stability of solution. | ||
Wavelength selection is hence critical to avoid bubble formation during bulk photopolymerization of an azide and alkyne based resin. Camphorquinone (5; CQ), a photosensitizing cyclic ketone, is a well-known visible light photoinitiator with peak absorption of 469 nm36 and is used in photopolymerization of methacrylate monomer-based dental composites.37 CQ alone has been shown to photoinitiate CuAAC using a mercury lamp equipped with a 400–500 nm band filter for small molecules by Tasdelen et al.38 and in bulk polymerization by Song et al.33 at moderately elevated temperatures (35 °C). Under blue-light during photopolymerization of vinyl monomers, the co-initiator amine species in conjunction with CQ has been utilized as a kinetic accelerator.39 Motivated by this approach, we developed a novel CQ–amine photoinitiation system for accelerated bulk photo-CuAAC polymerization networks.
The CQ–tertiary amine is a Norrish Type II photoinitiation system and follows a well-known two stage initiation process. Briefly, 470 nm wavelength light excites the CQ to a singlet state during the n–σ* transition. After an inter-system crossing (ISC) from the singlet to the triplet state, CQ interacts via electron transfer with the tertiary amine to form an excited complex (exciplex), which then extracts hydrogen from the amine to form a radical on the α-C atom of the tertiary amine.40,41 The stability of this radical is critical in the hydrogen transfer process.37,39,42 Unique to the photo-CuAAC reaction scheme, the radical on the amine then reduces Cu(II) to Cu(I) which in turn catalyzes the CuAAC reaction portrayed in Fig. 3A.
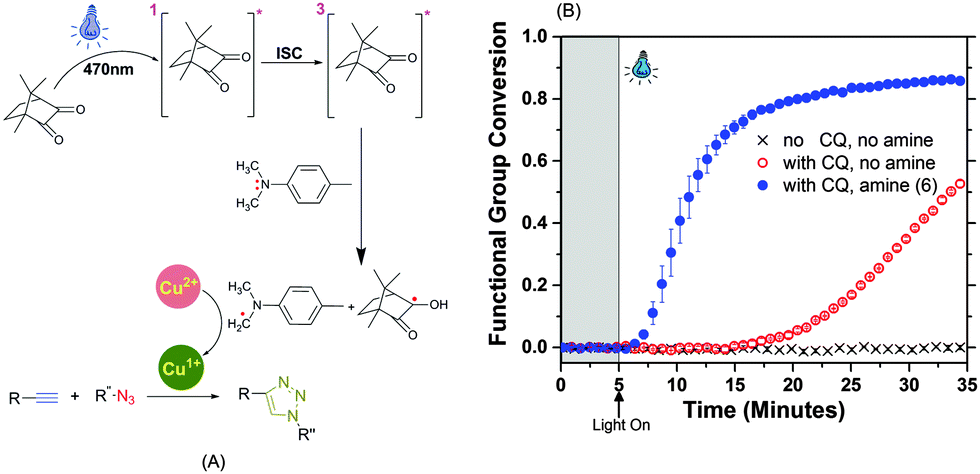 | ||
Fig. 3 (A) Camphorquinone-amine photoinitiation mechanism for photo-CuAAC. (B) The conversion of the alkyne functional group (6540–6460 cm−1) was tracked using real-time near infrared (NIR) spectroscopy during photopolymerization for monomer formulation containing 1:1 functional group ratio of azide:alkyne functional group, 1.5 wt% of 3, with or without presence of 0.35 wt% of 5 and 6, irradiated with 470 nm blue-light of 40 mW cm−2 after 5 minutes in dark (marked by ( ) shaded region). ) shaded region). | ||
The accelerated photopolymerization kinetics were monitored in real-time by tracking decrease in the area of the alkyne peak (6540–6460 cm−1) using near-IR (NIR) spectroscopy while simultaneously irradiating the sample (Fig. 3A). The inclusion of a tertiary amine in the CuAAC formulation yields a rapid alkyne functional group conversion of ≈80% after 15 minutes. The amine was added to monomers in 1:1 CQ:amine ratio (w/w) (i.e., 0.35 wt% of 5 and 0.35 wt% of 6 of the total monomer weight in the CuAAC formulation) and polymerized under 470 nm filtered light of intensity 40 mW cm−2 to yield a bubble free network. The formulations not containing the accelerating amine exhibit only 5% conversion at equal irradiation intensity and time; it is possible that the excited CQ could abstract an H-atom from the PMDETA (Cu-ligand)18,43 or from the urethane group on monomer 1.33,44 The control sample with no CQ and no amine shows negligible conversion throughout. The CQ:amine ratio used is optimized at various light intensities to ensure minimum leachable amine in the glassy network (see Fig. S2 in ESI†). The effect of light intensity, relative amounts of CQ and amine, and kinetics of three different amines are also studied in detail (see Fig. S3–S5 in ESI†).
One of the main attributes of photo-CuAAC reactions is the ability of the reaction to continue after the initiating light has ceased (i.e., dark-polymerization).19,32,33 In Fig. 4 the polymerization conversion is tracked as a function of irradiation durations (5, 10, 15 and 40 minutes). The rate and extent of the dark polymerization depends on initial time of visible light illumination converting the corresponding amount of Cu(II) to Cu(I). The kinetics of the polymerization are similar at all irradiation times, depicting its efficiency in forming a stable Cu(I) catalyst from the Type II photoinitiation system and retaining the dark polymerization behaviour that is characteristic of photo-CuAAC systems. Not only can the sample be irradiated at short time intervals while maintaining similar kinetic results, but the overall reaction also reaches high conversions in a short amount of time (>75% conversion in less than 10 minutes). The dark polymerization behaviour is seen with different amine structures in photo-CuAAC networks and upon tracking the reaction for 12 hours, the networks reaches about 98% conversion due to the stability of the Cu(I) catalyst in the network (see Fig. S6 in ESI†). The defect-free network films polymerized under blue-light are optically clear unlike the UV polymerized films which are rendered ineffective in applications requiring transparency (see Fig. S7 in ESI†).
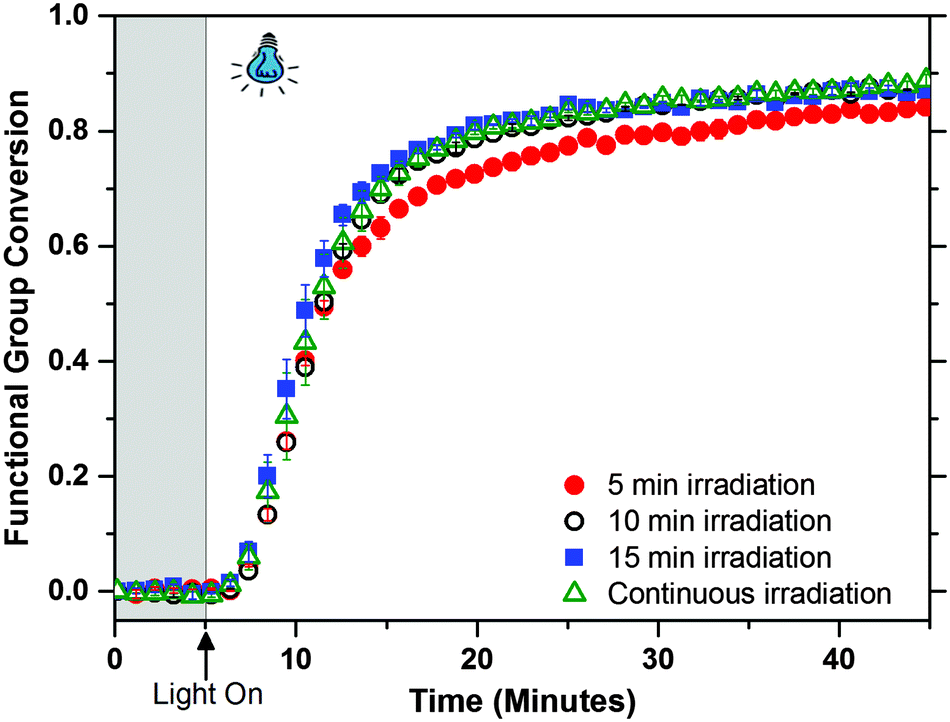 | ||
| Fig. 4 Real-time NIR spectroscopy for bulk photo-CuAAC reaction showing ‘dark polymerization’. The formulation contains 1:1 azide:alkyne functional groups, 1.5 wt% of 3, and 0.35 wt% of 5 and 6 each and was irradiated at intensity of 40 mW cm−2 with 470 nm blue-light. All samples were irradiated after 5 minutes but the specimens varied in terms of time under irradiation. All irradiation times exhibit similar final alkyne conversion of ≈89% after 40 minutes (5 minutes of irradiation yields a final conversion of ≈85%). | ||
The mechanical properties of the glassy, defect-free photo-CuAAC network sample (image in Fig. 5A), photopolymerized under ambient conditions are of prime importance. These properties are analysed using a Dynamic Mechanical Analyser (DMA) (shown in Fig. 5A). The narrow Tan Delta peak is indicative of a homogenous crosslinked structure formed via a step-growth polymerization mechanism The glass transition temperature (Tg) of 67 °C and the storage modulus of 2.2 GPa at room temperature makes photo-CuAAC conducive for glassy film applications. Furthermore, these results are consistent with those reported on photo-CuAAC networks formed using the same monomers with other photoinitiation systems.45
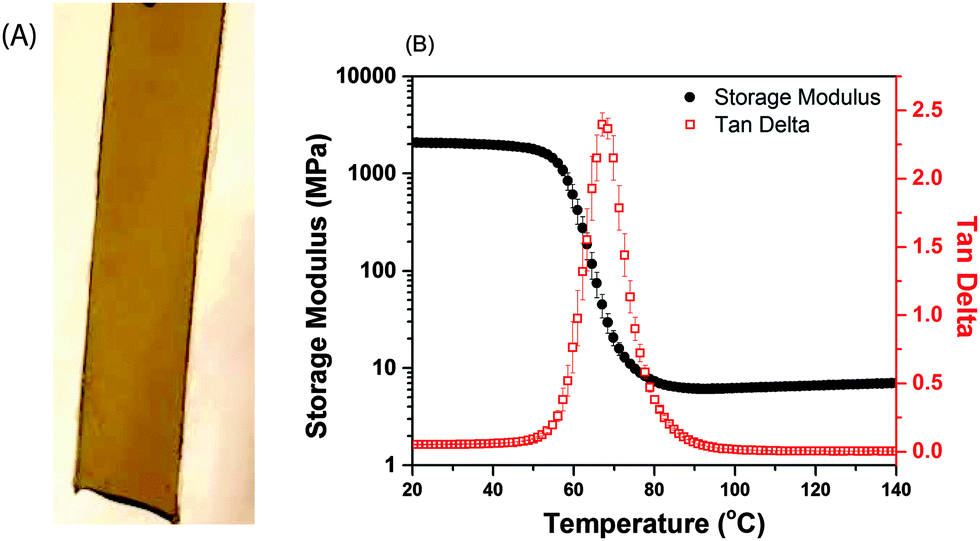 | ||
Fig. 5 (A) Defect-free photo-CuAAC sample. (B) Dynamic mechanical analysis of the defect-free photo-CuAAC sample. The storage modulus (●, 2.2 GPa) and Tan Delta ( , glass transition temperature Tg of 67 °C) of the polymer sample. Both (A) and (B) specimens are 0.5 mm thick, formulated using 1:1 azide:alkyne functional groups, 1.5 wt% of 3, and 0.35 wt% of 5 and 6 each and irradiated for 15 minutes under 470 nm blue-light at 40 mW cm−2 intensity. , glass transition temperature Tg of 67 °C) of the polymer sample. Both (A) and (B) specimens are 0.5 mm thick, formulated using 1:1 azide:alkyne functional groups, 1.5 wt% of 3, and 0.35 wt% of 5 and 6 each and irradiated for 15 minutes under 470 nm blue-light at 40 mW cm−2 intensity. | ||
In conclusion, a novel 470 nm (blue-light) initiation system is developed for photo-CuAAC bulk polymerizations. This readily integrable new method provides significant advantages over previous systems. First, azide decomposition is prevented and hence the polymer networks are transparent and defect-free. Second, the kinetics of photo-CuAAC are accelerated (>75% conversion in 10 minutes) at room temperature and optimized for a Type II photoinitiator system using camphorquinone in conjunction with a tertiary amine. Third, the network retains the characteristics that are desirable with photo-CuAAC systems, including dark polymerization after photoinitiation is ceased, high moduli, and narrow glass transition temperatures.
We acknowledge the financial support from NIH-NIDCR (U01 DE023774) and NIH-COBRE (1P30 GM110758). We thank Marco Dunwell, Jeffrey Heyes, and Prof. Bingjun Xu for help with gas-chromatographic measurements.
Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- W. X. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- Y. Kihara, T. Ichikawa, S. Abe, N. Nemoto, T. Ishihara, N. Hirano and M. Haruki, Polym. J., 2014, 46, 175–183 CrossRef CAS.
- N. W. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC.
- A. Noureddine, L. Lichon, M. Maynadier, M. Garcia, M. Gary-Bobo, J. I. Zink, X. Cattoen and M. Wong Chi Man, Nanoscale, 2015, 7, 11444–11452 RSC.
- D. Astruc, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630–640 CrossRef CAS PubMed.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7–17 CAS.
- S. Chatterjee and S. Ramakrishnan, Chem. Commun., 2013, 49, 11041–11043 RSC.
- S. C. Ritter and B. Konig, Chem. Commun., 2006, 4694–4696 RSC.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945–6947 CrossRef CAS.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
- C. N. Bowman, B. A. Adzima and C. J. Kloxin, US Pat., US9176380B2, 2015 Search PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.5b00586.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- F. S. Ekholm, H. Pynnonen, A. Vilkman, J. Koponen, J. Helin and T. Satomaa, Org. Biomol. Chem., 2016, 14, 849–852 CAS.
- N. P. Gritsan, Russ. Chem. Rev., 2007, 76, 1139–1160 CrossRef CAS.
- N. Gritsan and M. Platz, Organic azides: syntheses and applications, John Wiley, UK, 2010, ch. 11, pp. 311–364 Search PubMed.
- Computational methods in photochemistry, ed. A. G. Kutateladze, Taylor & Francis, Boca Raton, 2005 Search PubMed.
- G. L'Abbe, Chem. Rev., 1969, 69, 345–363 CrossRef.
- G. T. Hermanson, Bioconjugate techniques, Academic Press, San Diego, 3rd edn, 1996 Search PubMed.
- J. F. W. Keana and S. X. Cai, J. Org. Chem., 1990, 55, 3640–3647 CrossRef CAS.
- B. Howard, N. D. Wilson, S. M. Newman, C. S. Pfeifer and J. W. Stansbury, Acta Biomater., 2010, 6, 2053–2059 CrossRef CAS PubMed.
- C. S. Pfeifer, J. L. Ferracane, R. L. Sakaguchi and R. R. Braga, Am. J. Dent., 2009, 22, 206–210 Search PubMed.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 25, 2024–2028 CrossRef CAS PubMed.
- H. B. Song, A. Baranek and C. N. Bowman, Polym. Chem., 2016, 7, 603–612 RSC.
- M. K. McBride, T. Gong, D. P. Nair and C. N. Bowman, Polym. J., 2014, 55, 5880–5884 CAS.
- A. A. Alzahrani, D. P. Nair, D. J. Smits, M. Saed, C. M. Yakacki and C. N. Bowman, Chem. Mater., 2014, 26, 5303–5309 CrossRef CAS.
- Y. C. Chen, J. L. Ferracane and S. A. Prahl, Dent. Mater., 2007, 23, 655–664 CrossRef CAS PubMed.
- W. D. Cook, Polymer, 1992, 33, 600–609 CrossRef CAS.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45, 56–61 CrossRef CAS.
- W. F. Schroeder and C. I. Vallo, Dent. Mater., 2007, 23, 1313–1321 CrossRef CAS PubMed.
- A. Aguirre-Soto, A. T. Hwang, D. Gugla, J. W. Wydra, R. R. McLeod, C. N. Bowman and J. W. Stansbury, Macromolecules, 2015, 48, 6781–6790 CrossRef CAS.
- T. Y. Kwon, R. Bagheri, Y. K. Kim, K. H. Kim and M. F. Burrow, J Investig Clin Dent, 2012, 3, 3–16 CrossRef PubMed.
- W. Li, Y. Duan, M. Zhang, J. Cheng and C. Zhu, Chem. Commun., 2016, 52, 7596–7599 RSC.
- A. A. Alzahrani, A. H. Erbse and C. N. Bowman, Polym. Chem., 2014, 5, 1874–1882 RSC.
- S. Asmusen, G. Arenas, W. D. Cook and C. Vallo, Dent. Mater., 2009, 25, 1603–1611 CrossRef CAS PubMed.
- A. Baranek, H. B. Song, M. McBride, P. Finnegan and C. N. Bowman, Macromolecules, 2016, 49, 1191–1200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Gas-chromatography analysis of defect from monomer 1 under irradiation. The effect on kinetics from photo-initiator concentration optimization, light intensity, three different amine structure and overnight conversion showing dark polymerization. Effect of defects on clarity of photo-CuAAC network films. See DOI: 10.1039/c6cc05095f |
| This journal is © The Royal Society of Chemistry 2016 |